
Electron localization and the transition from adiabatic to nonadiabatic charge transport in organic conductors†
Abstract
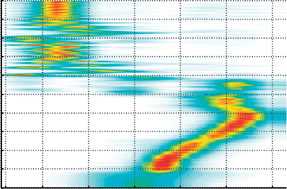
This tutorial review describes an atomistic simulation approach to studies of dynamics of charge
- This article is part of the themed collection: Conducting polymers for carbon electronics
 Please wait while we load your content...
Please wait while we load your content...

