
Stochastic approach to laser-induced ultrafast dynamics: the desorption of H2/D2 from Ru(0001)
Abstract
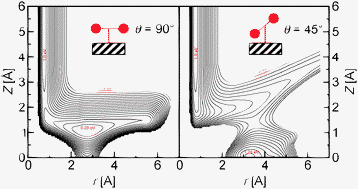
The desorption of molecular hydrogen and deuterium induced by femtosecond-laser pulses is studied theoretically for the so-called DIMET (Desorption Induced by Multiple Electronic Transitions) process. These investigations are based on nonadiabatic classical Monte Carlo trajectory (CMCT) simulations on a ground and an excited state potential energy surface, including up to all six adsorbate degrees of freedom. The focus is on the hot-electron mediated energy transfer from the surface to the molecule and back, and the energy partitioning between the different degrees of freedom of the desorbing molecules. We first validate for a two-mode model comprising the desorption mode and the internal vibrational coordinate, the classical Monte Carlo trajectory method by comparing with Monte Carlo wavepacket (MCWP) calculations arising from a fully quantum mechanical open-system density
 Please wait while we load your content...
Please wait while we load your content...

