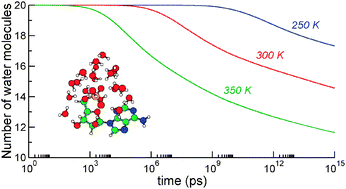
The structure and finite-temperature properties of hydrated nucleotide anion adenosine 5′-monophosphate (AMP) have been theoretically investigated with a variety of methods. Using a polarizable version of the Amber force field and replica-exchange molecular dynamics simulations, putative lowest-energy structures have been located for the AMP−(H2O)n cluster anions with n = 0–20. The hydration energies obtained with the molecular mechanics potential slightly overestimate experimental measurements. However, closer values are found after reoptimizing the structures locally at more sophisticated levels, namely semi-empirical (PM6) and density-functional theory (B3LYP/6-31+G*). Upon heating the complexes, various indicators such as the heat capacity, number of hydrogen bonds or surface area provide evidence that the water cluster melts below 200 K but remains bonded to the AMP anion. The sequential loss of water molecules after sudden heating has been studied using a statistical approach in which unimolecular evaporation is described using the orbiting transition state version of phase space theory, together with anharmonic densities of vibrational states. The evaporation rates are calibrated based on the results of molecular dynamics trajectories at high internal energy. Our results indicate that between 4 and 10 water molecules are lost from AMP−(H2O)20 after one second depending on the initial heating in the 250–350 K range, with a concomitant cooling of the remaining cluster by 75–150 K.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


