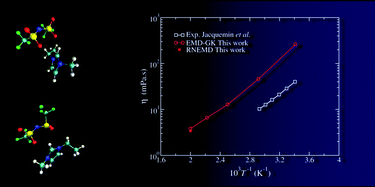
This work deals with the determination, using equilibrium and nonequilibrium molecular dynamics, of the viscosity of an ionic liquid: 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([emim][Tf2N]). A first method consists in computing the shear viscosity using the Green–Kubo formalism from the pressure tensor correlation function obtained from equilibrium simulations. On the other hand, the Newtonian viscosity can be extracted from the shear rate dependence of the viscosity in nonequilibrium simulations using the mode-coupling theory and the standard Cross and Carreau equations. We show that both methods lead to the same viscosity value, provided that the simulation time is long enough for the first method (much longer than advocated in the literature) and that special care is taken in the extrapolation procedure of the latter (simulations must be performed at very low shear rate). The force field employed in this work leads to an overestimation of the viscosity, with respect to experiment, by a factor ranging between 6 and 4 in the 293 K to 500 K temperature domain.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


