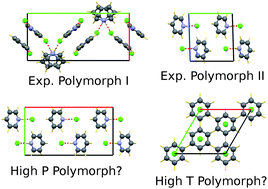
A crystal structure prediction study for the molecular salt pyridinium chloride was carried out with Z′ = 1 and Z′ = 2 in all 230 space groups. The predicted crystal structures were ranked by energy using dispersion-corrected density functional theory calculations, with the dispersion-correction parameterised against uncharged systems only. The experimental structures are ranked 1st and shared 2nd, which suggests that the dispersion-correction parameters are likely to be at least partially transferable to charged systems. For the structure generation step, a high-accuracy tailor-made force field was prepared for pyridinium chloride. The accuracy of the tailor-made force field is comparable to those for neutral molecules. A problem caused by spurious pseudo-symmetry due to the use of a cascade of energy potentials of different accuracies is described, as is its solution. Our calculations confirm that a previously reported P21/m structure for pyridinium chloride should have been reported in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , our calculations suggest the existence of a high-pressure polymorph in Pnma, and our calculations suggest that the high-temperature phase of pyridinium chloride might be subtly different from the high-temperature phase of pyridinium iodide.
, our calculations suggest the existence of a high-pressure polymorph in Pnma, and our calculations suggest that the high-temperature phase of pyridinium chloride might be subtly different from the high-temperature phase of pyridinium iodide.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


