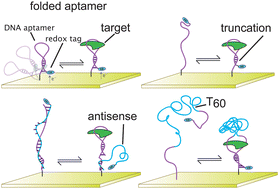
Electrochemical aptamer-based (E-AB) sensors have emerged as a promising and versatile new biosensor platform. Combining the generality and specificity of aptamer–ligand interactions with the selectivity and convenience of electrochemical readouts, this approach affords the detection of a wide variety of targets directly in complex, contaminant-ridden samples, such as whole blood, foodstuffs and crude soil extracts, without the need for exogenous reagents or washing steps. Signaling in this class of sensors is predicated on target-induced changes in the conformation of an electrode-bound probe aptamer that, in turn, changes the efficiency with which a covalently attached redox tag exchanges electrons with the interrogating electrode. Aptamer selection strategies, however, typically do not select for the conformation-switching architectures, and as such several approaches have been reported to date by which aptamers can be re-engineered such that they undergo the binding-induced switching required to support efficient E-AB signaling. Here, we systematically compare the merits of these re-engineering approaches using representative aptamers specific to the small molecule adenosine triphosphate and the protein human immunoglobulin E. We find that, while many aptamer architectures support E-AB signaling, the observed signal gain (relative change in signal upon target binding) varies by more than two orders of magnitude across the various constructs we have investigated (e.g., ranging from −10% to 200% for our ATP sensors). Optimization of the switching architecture is thus an important element in achieving maximum E-AB signal gain and we find that this optimal geometry is specific to the aptamer sequence upon which the sensor is built.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


