
Large-scale compensation of errors in pairwise-additive empirical force fields: comparison of AMBER intermolecular terms with rigorous DFT-SAPT calculations†
Abstract
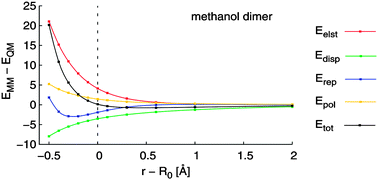
The intermolecular interaction energy components for several molecular complexes were calculated using force fields available in the AMBER suite of programs and compared with Density Functional Theory–Symmetry Adapted Perturbation Theory (DFT-SAPT) values. The extent to which such comparison is meaningful is discussed. The comparability is shown to depend strongly on the intermolecular distance, which means that comparisons made at one distance only are of limited value. At large distances the coulombic and van der Waals 1/r6 empirical terms correspond fairly well with the DFT-SAPT electrostatics and dispersion terms, respectively. At the onset of electronic overlap the empirical values deviate from the reference values considerably. However, the errors in the force fields tend to cancel out in a systematic manner at equilibrium distances. Thus, the overall performance of the force fields displays errors an order of magnitude smaller than those of the individual interaction energy components. The repulsive 1/r12 component of the van der Waals expression seems to be responsible for a significant part of the deviation of the force field results from the reference values. We suggest that further improvement of the force fields for intermolecular interactions would require replacement of the nonphysical 1/r12 term by an exponential function. Dispersion anisotropy and its effects are discussed. Our analysis is intended to show that although comparing the empirical and non-empirical interaction energy components is in general problematic, it might bring insights useful for the construction of new force fields. Our results are relevant to often performed force-field-based interaction energy decompositions.
 Please wait while we load your content...
Please wait while we load your content...

