
Study of the interaction between short alkanethiols from ab initio calculations
Abstract
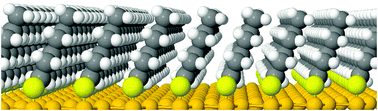
We have developed a force field to describe the interaction of alkanethiols HS(CH2)n−1 CH3 (Cn for short) by fitting a set of ∼220 interaction energies for dimers of Cn (with n = 1,2,…6) and CH4 molecules obtained from second-order Møller–Plesset perturbation theory calculations. The derived force field, based on a sum of so-called exp-6 pairwise potentials and effective Coulomb interaction potential between the HS- heads, predicts very well the interaction energies for dimers and trimers of alkanethiols not included in the input database for the fit. Also the calculated minimum energy tilt angle of the alkyl chains for a hexagonal arrangement of alkanethiolates with a nearest neighbor distance of 5 Å is in good agreement with the available 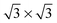 self-assembled monolayer (SAM) on
self-assembled monolayer (SAM) on
 Please wait while we load your content...
Please wait while we load your content...

