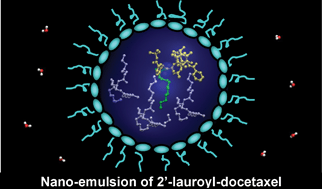
Computer-based theoretical calculations were employed to direct the design of docetaxel conjugates with enhanced solubility in the internal phase of a nano-emulsion formulation. The theoretically-identified optimal docetaxel conjugates were synthesized by direct attachment of lauroyl moieties through an ester linkage to docetaxel. In comparison to docetaxel, the conjugates exhibited significantly improved solubility in oil, as predicted by our theoretical calculations. This contributed to high drug entrapment efficiencies (up to 97%) and a high drug loading capacity (5.7% w/w) for the docetaxel conjugates. The mono-substitution of an acyl group at C-2′ of docetaxel resulted in a conjugate with 37- to 46-fold lower cytotoxicity than that of the parent drug in two human cancer cell lines. Importantly, the activity exerted by the mono-substituted docetaxel on the cancer cells was due in part to the cytotoxicity of the parent drug that was released via hydrolysis of the ester bond between the lauroyl moiety and the drug under biologically relevant conditions. In contrast, di- and tri-substitution of acyl groups at C-2′, C-7 and/or C-10 of docetaxel resulted in non-hydrolysable conjugates that were found to be inactive. Overall, our results show that computer-based theoretical calculation is a promising strategy for guiding the enhancement of material–drug compatibility in formulation development. Also, these studies confirm that chemical modification of docetaxel for enhancement of material–drug compatibility should be limited to mono-substitution at C-2′ and result in a prodrug that is hydrolysable at a moderate rate under biologically relevant conditions.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


