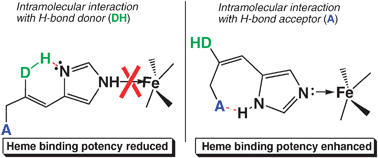
Enzymes involved in the mammalian microsomal metabolism of drugs are, in numerous cases, inhibited by compounds bearing an imidazolyl scaffold. However, the inhibition potency is highly dependent upon the accessibility of the imidazolyl nitrogen lone pair. In order to highlight some structural parameters of inhibitors that control this phenomenon, a series of compounds containing a nitrogen unsubstituted imidazolyl moiety with varying degrees of nitrogen lone pair accessibility was tested on human and rat hepatic cytochromes P450 and microperoxidase 8, an enzymatically active peptide derived from cytochrome c. In each case, we have shown that the accessibility of the imidazole lone pair determined the extent of inhibition. Nitrogen accessibility was tuned not only by varying the steric hindrance in the vicinity of the imidazolyl ring but also by modifying its surrounding hydrogen bonding network. Compounds in which there exists intramolecular hydrogen bonding between the imidazole moiety and an H-bond acceptor, such as an appropriately positioned amidecarbonyl group, demonstrated enhanced inhibitory effects. Conversely, imidazole moieties which are in proximity to H-bond donors, such as an amide NH group, displayed reduced potency. This trend was observed in cyclo-peptide derivatives in which the intramolecular H-bond network was adjusted through the modification of the stereochemistry of a dehydrohistidine residue. It was observed that (Z)-isomers weakly bind heme, whereas (E)-isomers demonstrated higher degrees of metal binding. Therefore, enzymatic inhibition of heme-containing proteins by compounds bearing a dehydrohistidine motif seems to be closely related to its stereochemistry and hydrogen binding propensity. At neutral pH, these differences in binding affinities can be confidently attributed to the ambident H-bond properties of imidazole nitrogen atoms. This structure-activity relationship may be of use for the design of novel imidazolyl compounds as new P450 inhibitors or drug candidates.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
