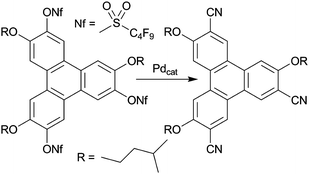
Despite recent advances in the application of discotic liquid crystals (DLC) in organic electronic devices a better understanding of structure–property relationships is required for designing commercially viable discotic semiconductors. This is a complex task because a single structural change to the DLC may affect many relevant properties of the discotic material such as chemical stability, mesomorphism, alignment, and electronic properties. Presented here is a comprehensive study of hexa-substituted triphenylene derivatives containing pentyloxy and 3-methylbutyloxy groups as well as one or three nonaflate or cyano groups. The compounds are synthesized by both established and new synthetic approaches and are isolated as pure isomers. Their structure–mesomorphism relationship is studied by optical polarized microscopy, thermal analysis, and variable temperature X-ray diffraction and their relationships between structure and frontier orbital energies are investigated by cyclic voltammetry in solution, UV-Vis spectroscopy and computational studies at the DFT level. Both mesomorphism and frontier orbital energies of the compounds depend on the number and positions of nonaflate and cyano groups while only their mesomorphism is affected by a change from linear pentyloxy to branched 3-methylbutyloxy chains. To our surprise lowering the symmetry of the compounds is equally effective in lowering HOMO–LUMO gaps than the attachment of strongly π-electron withdrawing cyano groups. Consequently, the non-symmetrically substituted triphenylenes with three cyano groups are the most electron deficient and have the smallest HOMO–LUMO gap of the compounds prepared here but their ELUMO of about −2.8 eV is still high in comparison to typical electron acceptors (ELUMO below −3.5 eV). The computational approach established for the prepared compounds is also employed for the prediction of frontier orbital energies of new electron deficient discotic triphenylene derivatives.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
