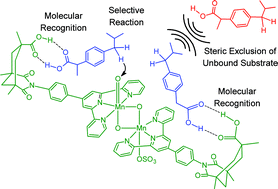
Experimental studies have shown that the C–H oxidation of Ibuprofen and methylcyclohexane acetic acid can be carried out with high selectivities using [(terpy’)Mn(OH2)(μ-O)2Mn(OH2)(terpy’)]3+ as catalyst, where terpy’ is a terpyridine ligand functionalized with a phenylene linker and a Kemp's triacid serving to recognize the reactant viaH-bonding. Experiments, described here, suggest that the sulfate counter anion, present in stochiometric amounts, coordinates to manganese in place of water. DFT calculations have been carried out using [(terpy’)Mn(O)(μ-O)2Mn(SO4)(terpy’)]+ as a model catalyst, to analyze the origin of selectivity and its relation to molecular recognition, as well as the mechanism of catalyst inhibition by tert-butyl benzoic acid. The calculations show that a number of spin states, all having radical oxygen character, are energetically accessible. All these spin states promote C–H oxidationvia a rebound mechanism. The catalyst recognizes the substrate by a double H bond. This interaction orients the substrate inducing highly selective C–H oxidation. The double hydrogen bond stabilizes the reactant, the transition state and the product to the same extent. Consequently, the reaction occurs at lower energy than without molecular recognition. The association of the catalyst with tert-butyl benzoic acid is shown to shield the access of unbound substrate to the reactive oxo site, hence preventing non-selective hydroxylation. It is shown that the two recognition sites of the catalyst can be used in a cooperative manner to control the access to the reactive centre.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


