
Structural evolution and properties of subnanometer Tcn (n = 2–15) clusters
Abstract
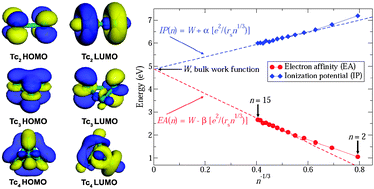
All-electron scalar relativistic calculations of the geometric and electronic structures of neutral subnanometer Tcn (n = 2–15) clusters have been performed using spin-polarized density functional theory. It is shown that the structural evolution of Tcnclusters from n = 3 to n = 8 is similar to Mn clusters, which also belong to the group of VIIB transition metals. The calculated binding energy per atom varies gradually from 0.65 eV for Tc2 to 2.35 eV for Tc15. The computed electron affinity and ionization potential follow the classical electrostatic metallic droplet model for clusters larger than Tc6 and converge asymptotically to the experimental value of the bulk Tc work function. A study of the kinetic stability of Tc clusters in terms of the second difference of binding energies and the energy gap between the frontier orbital states predicts Tc3(D3h), Tc4(Td), Tc6(Oh), Tc12(Ih), and Tc13(Ih) to be the most stable clusters.
 Please wait while we load your content...
Please wait while we load your content...

