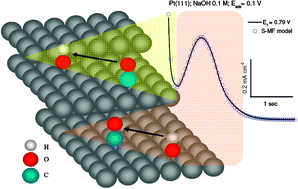
From a detailed analysis of the chronoamperometry of CO stripping on stepped platinum single-crystal electrodes in alkaline solution, in combination with kinetic modeling, a mechanistic and kinetic picture of the CO oxidation mechanism is derived. On Pt(111), CO oxidation starts at defect sites (steps, kinks), following a one-dimensional nucleation-and-growth mechanism, or a Langmuir–Hinshelwood mechanism with no effective competition between CO and OH. The carbonate that is formed in this reaction blocks the active oxidation sites, so that CO adsorbed on terraces further away from the defect sites can be oxidized at defects sites only very slowly. At potentials above ca. 0.75 V vs. RHE this CO is oxidized on the Pt(111) terrace. On stepped Pt electrodes, CO oxidation is also initiated at the kink (step defects) and step sites. We postulate that carbonate partially blocks the active site, but CO from the terrace still prefers to react at the steps over the terrace sites. The oxidation of terrace-bound CO at the step sites follows a competitive Langmuir–Hinshelwood mechanism. The least reactive CO on the surface is the CO that is adsorbed on the step sites, and it is oxidized by OH on terraces. Finally, on the terrace, the Tafel slope for CO stripping is close to 60 mV dec−1, suggesting an EC mechanism, i.e. reversible OH formation followed by a chemical rate-determining step, presumably the CO + OH combination reaction. At the step site, the Tafel slope is close to 120 mV dec−1, suggesting an electrochemical rate-determining step, ascribed to slow OH formation at the step site.

 Please wait while we load your content...
Please wait while we load your content...

