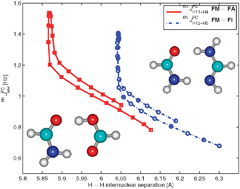
In this paper, we present density functional theory calculations to predict the NMR parameters for two model systems: the formamide–formic acid (FM⋯FA) and formamide–formamidine (FM⋯FI) complexes, where intermolecular double proton exchange occurs. For the first time, the NMR parameters have been calculated along the reaction paths of the proton transfers described by means of the intrinsic reaction coordinate (IRC) procedure. The most interesting one-bond spin–spin coupling constants, 1(h)JXH, between migrating protons and heavier nuclei change character from intra- to intermolecular along the pathway. The maximal positive values of the reduced 1(h)KXH coupling constants correspond to the situation when they are intramolecular; they decrease along the path, change sign and reach small negative values, becoming intermolecular couplings. The differing character of the double proton exchange resulting from the synchronicity or asynchronicity of the process is reflected in the calculated NMR parameters. Surprisingly substantial values have been calculated for the six-bond intermolecular proton–proton6hJHH coupling constants between protons bound to the carbon atoms. A simple procedure consisting of removal of the proton(s) forming the hydrogen bonds has been employed to indicate an influence of hydrogen bonding on the intermolecular coupling constants. Some of the spin–spin coupling constants (2hJXY) are predominantly transmitted through hydrogen bonds and decrease with removal of the proton(s), while others (4hJCC) are less sensitive to the presence or absence of the protons of hydrogen bonding.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


