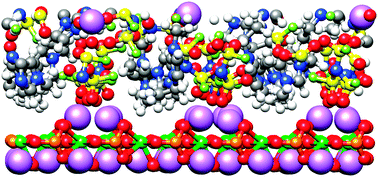
We have performed atomistic molecular dynamics (MD) simulations of the (010) surface of LiFePO4 in contact with an organic liquid electrolyte (OLE), ethylene carbonate : dimethyl carbonate (3 : 7) with approximately 1 mol kg−1 LiPF6, and an ionic liquid-based electrolyte (ILE), 1-ethyl 3-methyl-imidazolium: bis(fluorosulfonyl)imide (EMIM+ : FSI−) with approximately 1 mol kg−1LiFSI. Surface-induced structure that extends about 1 nm from the LiFePO4 surface was observed in both electrolytes. The electrostatic potential at the LiFePO4 surface was found to be negative relative to the bulk electrolyte reflecting an excess of negative charge from the electrolyte coordinating surface Li+. In the ILE system negative surface charge is partially offset by a high density of EMIM+ cations coordinating surface oxygen. The electrostatic potential exhibits a (positive) maximum about 3 Å from the LiFePO4 surface which, when combined with the reduced ability of the highly structured electrolytes to solvate Li+ cations, results in a free energy barrier of almost 4 kcal mol−1 for penetration of the interfacial electrolyte layer by Li+. The resistance for bringing Li+ from the bulk electrolyte to the LiFePO4 surface through this interfacial barrier was found to be small for both the OLE and ILE. However, we find that the ability of EMIM+ cations to donate positive charge to LiFePO4/electrolyte interface may result in a significant decrease in the concentration of Li+ at the surface and a corresponding increase in impedance to Li+ intercalation into LiFePO4, particularly at lower temperatures.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


