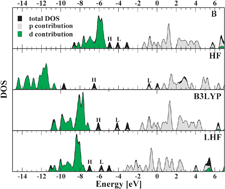
We assess the localized Hartree–Fock (LHF) method for the simulation of the structural and electronic properties of gold microclusters. We compute the minimum-energy geometries, atomization energies, dipole moments, and single-particle spectra for five gold clusters, AuN, and their anions, AuN−, with N≤ 4. Calculations are performed with the LHF functional, with and without the addition of the Lee–Yang–Parr (LYP) correlation, and the results are compared with those obtained from other semilocal and hybrid functionals as well as from Hartree–Fock calculations. The LHF functional yields structural properties of similar quality to the other exchange-only methods, but superior molecular orbital energies are found. The LHF single-particle spectrum is free of the artifacts typical for HF and standard DFT calculations and in good agreement with available experimental reference data. The inclusion of correlation by the LYP functional produces a significant improvement of geometries and energetics, but has only a minor impact on the orbital energies.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


