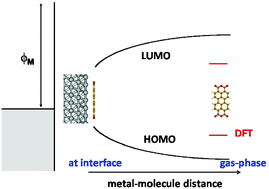
A review of our theoretical understanding of the band alignment at organic interfaces is presented with particular emphasis on the metal/organic (MO) case. The unified IDIS (induced density of interface states) and the ICT (integer charge transfer) models are reviewed and shown to describe qualitatively and semiquantitatively the barrier height formation at those interfaces. The IDIS model, governed by the organic CNL (charge neutrality level) and the interface screening includes: (a) charge transfer across the interface; (b) the “pillow” (or Pauli) effect associated with the compression of the metal wavefunction tails; and (c) the molecular dipoles. We argue that the ICT-model can be described as a limiting case of the unified IDIS-model for weak interface screening. For a fully quantitative understanding of the band alignment at organic interfaces, use of DFT (density functional theory) or quantum chemistry methods is highly desirable. In this Perspective review, we concentrate our discussion on DFT and show that conventional LDA or GGA calculations are limited by the “energy gap problem of the organic materials”, because the LDA (or GGA) Kohn–Sham energy levels have to be corrected by the self-interaction energy of the corresponding wavefunction, to provide the appropriate molecule transport energy gap. Image potential and polarization effects at MO interfaces tend to cancel these self-interaction corrections; in particular, we show that for organic molecules lying flat on Cu and Ag, these cancellations are so strong that we can rely on conventional DFT to calculate their interface properties. For Au, however, the cancellations are weaker making it necessary to go beyond conventional DFT. We discuss several alternatives beyond conventional LDA or GGA. The most accurate approach is the well-known GW-technique, but its use is limited by its high demanding computer time. In a very simple approach one can combine conventional DFT with a “scissor” operator which incorporates self-interaction corrections and polarization effects in the organic energy levels. Hybrid potentials combined with conventional DFT represent, probably, the best alternative for having a simple and accurate approach for analyzing organic interfaces. The problem then is to find an appropriate one for both the metal and the organic material in a plane-wave formulation; we show, however, how to overcome this difficulty using a local-orbital basis formulation. As examples of these alternatives, we present some DFT-calculations for several organic interfaces, using either the scissor operator or a hybrid potential, which can be interpreted in terms of the unified IDIS-model.

 Please wait while we load your content...
Please wait while we load your content...

