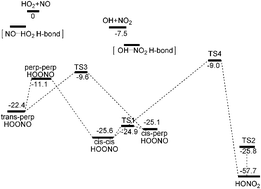
We report quasiclassical trajectory calculations of the HO2 + NO reaction using a new full dimensional, singlet potential energy surface (PES) which is a fit to more than 67 000 energies obtained with density functional theory-B3LYP/6-311G(d,p)-calculations. The PES is invariant with respect to permutation of like nuclei and describes all isomers of HOONO, HONO2, saddle points connecting them and the OH + NO2, HO2 + NO channels. Quasiclassical trajectory calculations of cross-sections for the HO2 + NO to form HOONO, HONO2 and OH + NO2 are done using this PES, for reactants in the ground vibrational state and rotational states sampled from a 300 K Boltzmann distribution. Trajectory calculations illustrate the pathway that HO2 + NO takes to the energized HOONO complex, which dissociates to products OH + NO2, reactants HO2 + NO, or isomerizes to HONO2. The association cross sections are used to obtain rate constants for formation of HOONO and HONO2 in the high-pressure limit, and formation of products OH + NO2 in the low-pressure limit.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


