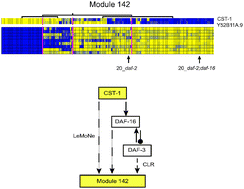
Differential gene expression governs the development, function and pathology of multicellular organisms. Transcription regulatory networks study differential gene expression at a systems level by mapping the interactions between regulatory proteins and target genes. While microarray transcription profiles are the most abundant data for gene expression, it remains challenging to correctly infer the underlying transcription regulatory networks. The reverse-engineering algorithm LeMoNe (learning module networks) uses gene expression profiles to extract ensemble transcription regulatory networks of coexpression modules and their prioritized regulators. Here we apply LeMoNe to a compendium of microarray studies of the worm Caenorhabditis elegans. We obtain 248 modules with a regulation program for 5020 genes and 426 regulators and a total of 24 012 predicted transcription regulatory interactions. Through GO enrichment analysis, comparison with the gene–gene association network WormNet and integration of other biological data, we show that LeMoNe identifies functionally coherent coexpression modules and prioritizes regulators that relate to similar biological processes as the module genes. Furthermore, we can predict new functional relationships for uncharacterized genes and regulators. Based on modules involved in molting, meiosis and oogenesis, ciliated sensory neurons and mitochondrial metabolism, we illustrate the value of LeMoNe as a biological hypothesis generator for differential gene expression in greater detail. In conclusion, through reverse-engineering of C. elegans expression data, we obtained transcription regulatory networks that can provide further insight into metazoan development.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
