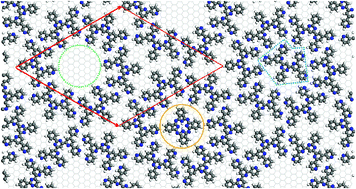
Host–guest networks formed by ordered organic layers are promising candidates for applications in molecular storage and quantum computing. We have studied 2-dimensionally ordered surface template structures of bis(terpyridine)-derived molecules (BTPs) on graphite using force field and DFT methods and compared the results to recent experimental observations. In order to determine the force field best suited for surface calculations, bond lengths and angles, torsional potentials, adsorption and stacking energies of smaller aromatic molecules were calculated with different force fields (Compass, UFF, Dreiding and CVFF). Density functional perturbation theory calculations were used to study the intermolecular interactions between 3,3′-BTP molecules. Structural properties, adsorption energies and rotational barriers of the 3,3′-BTP surface structure and its host–guest systems with phthalocyanine (PcH2) or excess 3,3′-BTP as guest molecules have been addressed. In addition, STM images of oligopyridine and phthalocyanine molecules were simulated based on periodic and local density functional theory calculations.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


