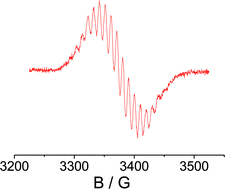
The radical complex {(μ4-TCNQF4)[Re(CO)3(bpy)]4}(PF6)3, as prepared and isolated from the reaction between TCNQF4 and [Re(CO)3(bpy)(MeOH)](PF6), was studied electrochemically and by IR vibrational spectroscopy, UV-Vis-NIR absorption spectroscopy, and by EPR at 9.5, 190 and 285 GHz. The isotropic g factor of 2.0058, the detectable g anisotropy, and the 185,187Re EPR hyperfine coupling of 0.95 mT for four equivalent metal nuclei support predominant, but not exclusive, spin localisation at the bridging ligand. Nitrile and metal carbonyl stretching frequencies as well as the typically structured near infrared absorption band lend further support to (TCNQF4˙−)(ReI)4 as the most appropriate oxidation state formulation. In comparison to the non-radical complex {(μ4-TCNQ)[Re(CO)3(bpy)]4}(PF6)4 an X-ray structure analysis of {(μ4-TCNQF4)[Re(CO)3(bpy)]4}(PF6)3 shows a marginally more twisted (ReNCCCNRe)(C6X4)(ReNCCCNRe) configuration and a different up/down arrangement of the [Re(CO)3(bpy)]+ groups. This first isolation, electrochemical, structural and spectroscopic characterisation of a discrete tetranuclear radical complex of a TCNQ-type ligand suggests a link between the stability of such materials and the rather small structural changes on ligand-based electron transfer.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


