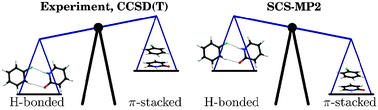
Fluorobenzenes are π-acceptor synthons that form π-stacked structures in molecular crystals as well as in artificial DNAs. We investigate the competition between hydrogen bonding and π-stacking in dimers consisting of the nucleobase mimic 2-pyridone (2PY) and all fluorobenzenes from 1-fluorobenzene to hexafluorobenzene (n-FB, with n = 1–6). We contrast the results of high level ab initio calculations with those obtained using ultraviolet (UV) and infrared (IR) laser spectroscopy of isolated and supersonically cooled dimers. The 2PY·n-FB complexes with n = 1–5 prefer double hydrogen bonding over π-stacking, as diagnosed from the UV absorption and IR laser depletion spectra, which both show features characteristic of doubly H-bonded complexes. The 2-pyridone·hexafluorobenzene dimer is the only π-stacked dimer, exhibiting a homogeneously broadened UV spectrum and no IR bands characteristic for H-bonded species. MP2 (second-order Møller–Plesset perturbation theory) calculations overestimate the π-stacked dimer binding energies by about 10 kJ/mol and disagree with the experimental observations. In contrast, the MP2 treatment of the H-bonded dimers appears to be quite accurate. Grimme’s spin-component-scaled MP2 approach (SCS-MP2) is an improvement over MP2 for the π-stacked dimers, reducing the binding energy by ∼10 kJ/mol. When applied to explicitly correlated MP2 theory (SCS-MP2-R12 approach), agreement with the corresponding coupled-cluster binding energies [at the CCSD(T) level] is very good for the π-stacked dimers, within ±1 kJ/mol for the 2PY complexes with 1-fluorobenzene, 1,2-difluorobenzene, 1,2,4,5-tetrafluorobenzene, pentafluorobenzene and hexafluorobenzene. Unfortunately, the SCS-MP2 approach also reduces the binding energy of the H-bonded species, leading to disagreement with both coupled-cluster theory and experiment. The SCS-MP2-R12 binding energies follow the SCS-MP2 binding energies closely, being about 0.5 and 0.7 kJ/mol larger for the H-bonded and π-stacked forms, respectively, in an augmented correlation-consistent polarized valence quadruple-zeta basis. It seems that the SCS-MP2 and SCS-MP2-R12 methods cannot provide sufficient accuracy to replace the CCSD(T) method for intermolecular interactions where H-bonding and π-stacking are competitive.

 Please wait while we load your content...
Please wait while we load your content...

