
A first-principles study of K adsorption on Pb(111)
Abstract
Ab
initio total-energy density functional theory calculations with supercell models have been employed to investigate the 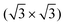 R30° and (2 × 2) structures of K on the Pb(111) surface. Four “on-surface” sites and a substitutional site were considered. The calculations showed that the substitutional site is more stable than all the on-surface sites, due to its low vacancy formation energy. The calculated
R30° and (2 × 2) structures of K on the Pb(111) surface. Four “on-surface” sites and a substitutional site were considered. The calculations showed that the substitutional site is more stable than all the on-surface sites, due to its low vacancy formation energy. The calculated 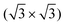 R30° geometry agrees well with the
R30° geometry agrees well with the

 Please wait while we load your content...
Please wait while we load your content...

