
Accurate calculations of intermolecular interaction energies using explicitly correlated wave functions
Abstract
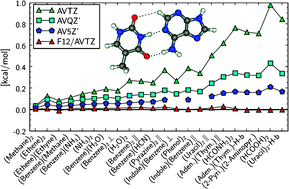
Explicitly correlated second-order Møller–Plesset (MP2-F12) calculations of intermolecular interaction energies for the S22 benchmark set of Jurečka, Šponer, Černý, and Hobza (Chem. Phys. Phys. Chem. 2006, 8, 1985) are presented and compared with standard MP2 results. The MP2 complete basis set limits are estimated using basis set extrapolation and augmented quadruple-zeta and quintuple-zeta basis sets. Already with augmented double-zeta basis sets the MP2-F12 interaction energies are found to be closer to the complete basis set limits than standard MP2 calculations with augmented quintuple-zeta basis sets. Various possible approximations in the MP2-F12 method are systematically tested. Best results are obtained with localized orbitals and the diagonal MP2-F12/C(D) ansatz. Hybrid approximations, in which some contributions of the auxiliary basis set are neglected and which considerably reduce the computational cost, have a negligible effect on the interaction energies. Also the orbital-invariant fixed-amplitude approximation of Ten-no leads to only slightly less accurate results. Preliminary results for the neon and
- This article is part of the themed collection: Explicit-r12 correlation methods and local correlation methods
 Please wait while we load your content...
Please wait while we load your content...

