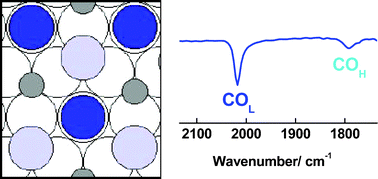
The dynamics of adsorption and oxidation of CO on Ru(0001) electrode in sulfuric acid solution have been studied using in situ FTIR spectroscopy under potential control and at open circuit, the latter at 20 and 55 °C. The in situ IR data show clearly that the bisulfate anion adsorbs on the Ru(0001) surface over the potential range from −200 mV to 350 mV (vs. Ag/AgCl) at 20 °C in the absence and presence of adsorbed CO; however, increasing the temperature to 55 °C and/or increasing the concentration of dissolved O2 reduces the bisulfate adsorption. The formation of surface (hydro-) oxide at higher potentials replaces the bisulfate adsorbates. Both linear (COL) and three-fold hollow bonded CO (COH) adsorbates were produced following CO adsorption at Ru(0001) in H2SO4, as was observed in our previous studies in HClO4. However, the amount of adsorbed CO observed in H2SO4 was ca. 10% less than that in HClO4; in addition, the COL and COH frequencies were higher in H2SO4, and the onset potential for COads oxidation 25 mV lower. These new results are interpreted in terms of a model in which the adsorbed bisulfate weakens the CO adlayer, allowing the active Ru oxide layer to form at lower potentials. Significantly different results were observed at open circuit in H2SO4 compared both to the data under potential control and to our earlier data in HClO4, and these observations were rationalized in terms of the adsorbed HSO4− anions (pre-adsorbed at −200 mV) inhibiting the oxidation of the surface at open circuit (after stepping from the initial potential of −200 mV), as the latter was no longer driven by the imposed electrochemical potential but via chemical oxidation by trace dissolved O2. Results from experiments at open circuit at 55 °C and using oxygen-saturated H2SO4 supported this model. The difference in Ru surface chemistry between imposed electrochemical control and chemical control has potential implications with respect to fuel cell electrocatalysis.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


