
Subsolidus phase relations in Ca2Mo2O8–NaEuMo2O8-powellite solid solution predicted from static lattice energy calculations and Monte Carlo simulations
Abstract
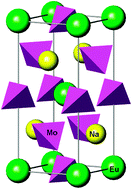
Thermodynamic ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) symmetry. The calculated subsolidus temperature-composition phase diagram is dominated by three miscibility gaps which are separated by narrow fields of stability of two ordered phases with the compositions of x = 4/9 and x = 2/3, where x is the mole fraction of the NaEuMo2O8 end-member.
symmetry. The calculated subsolidus temperature-composition phase diagram is dominated by three miscibility gaps which are separated by narrow fields of stability of two ordered phases with the compositions of x = 4/9 and x = 2/3, where x is the mole fraction of the NaEuMo2O8 end-member.
 Please wait while we load your content...
Please wait while we load your content...

