
Bicyclic proline analogues as organocatalysts for stereoselective aldol reactions: an in silico DFT study†
Abstract
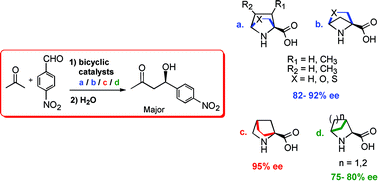
Density functional theory has been employed in investigating the efficiency of a series of bicyclic analogues of proline as stereoselective organocatalysts for the aldol reaction. Three classes of conformationally restricted proline analogues, as part of either a [2.2.1] or [2.1.1] bicyclic framework, have been studied. Transition states for the stereoselective C–C bond formation between enamines derived from [2.2.1] and [2.1.1] bicyclic amino acids and p-nitrobenzaldehyde, leading to enantiomeric products, have been identified. Analysis of the transition state geometries revealed that the structural rigidity of catalysts, improved transition state organization as well as other weak interactions influence the relative stabilities of diastereomeric transition states and help contribute to the overall stereoselectivity in the aldol reaction. These bicyclic catalysts are predicted to be substantially more effective in improving the enantiomeric excess than the widely used organocatalyst proline. Enantiomeric excesses in the range 82–95% are predicted for these bicyclic catalysts when a sterically unbiased substrate such as p-nitrobenzaldehyde is employed for the asymmetric aldol reaction. More interestingly, introduction of substituents, as simple as a methyl group, at the ortho position of the aryl aldehyde bring about an increase in the enantiomeric excess to values greater than 98%. The reasons behind the vital energy separation between diastereomeric transition states has been rationalized with the help of a number of weak interactions such as intramolecular hydrogen bonding and Coulombic interactions operating on the transition states. These predictions could have wider implications for the rational design of improved organocatalysts for stereoselective carbon–carbon bond-forming reactions.
 Please wait while we load your content...
Please wait while we load your content...

