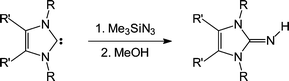
The preparation of 2-iminoimidazolines 3a–3f has been accomplished by the Staudinger reaction of the carbenes 1,3-di-tert-butylimidazolin-2-ylidene (1a), 1,3-diisopropyl-4,5-dimethylimidazolin-2-ylidene (1b), 1,3-diisopropylimidazolin-2-ylidene (1c), 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene (1d), 1,3-bis(2,6-diisopropylphenylimidazolin-2-ylidene (1e) and 1,3,4,5-tetramethylimidazolin-2-ylidene (1f) with trimethylsilyl azide (Me3SiN3) followed by desilylation of the resulting 2-trimethylsilyliminoimidazolines 2a–2e. The X-ray crystal structures of 2d and 2e have been established, revealing C1–N1–Si1 angles that are more obtuse than the corresponding P–N–Si angles observed in related trimethylsilyl iminophosphoranes. Together with 2c, the disilylated side product 1,3-diisopropyl-2-(trimethylsilylimino)-4-trimethylsilylimidazoline (4) has been isolated and structurally characterized. Cleavage of the N–Si bonds in 2a–2f and formation of 3a–3f is easily achieved by stirring in methanol. The molecular structures of the 2-iminoimidazolines 2a–2c are reported, indicating that the structural parameters are best described by non-ylidic resonance structures and that electron delocalization within the imidazole heterocycle does not play a crucial role in these imine systems. Compound 2a forms a head-to-head dimer in the solid state via weak intermolecular N–H⋯N contacts, which have additionally been characterized by means of compliance constants. To further analyze the electronic structure of these imines in comparison to related guanidine ligands, the proton affinities (PAs) of the model compounds 2-imino-1,3-dimethylimidazoline (5), 2-imino-1,3-dimethylimidazolidine (6) and tetramethylguanidine (7) have been calculated by means of density functional theory. Finally, the charge distribution in 5–7 and the relative contribution of relevant resonance structures have been determined using natural bond orbitals (NBO) and natural resonance theory (NRT).

 Please wait while we load your content...
Please wait while we load your content...

