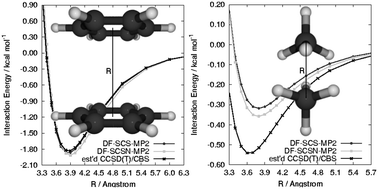
An examination of the performance of density-fitted, spin-component-scaled, second-order Møller–Plesset theory (SCS-MP2), SCS-MP2 with parameters optimized for nucleic acids (SCSN-MP2), and their local-correlation variants, SCS-LMP2 and SCSN-LMP2, is presented for the sandwich and T-shaped benzene dimers, the methane–benzene and H2S–benzene complexes, and the methane dimer over entire potential energy curves. These are compared to benchmark-quality estimates of the complete-basis-set limit for coupled-cluster theory through perturbative triple excitations, CCSD(T)/CBS. With the exception of the methane dimer, SCSN-LMP2/CBS tends to outperform SCS-LMP2/CBS with maximum relative errors of 6 and 18%, respectively, at the optimal CCSD(T)/CBS intermolecular distances. For the methane dimer, errors for SCS(N)-(L)MP2/CBS remain in the 0.2–0.3 kcal mol−1 range, corresponding to a larger relative error of 40–50%. Although the local MP2 methods perform very similarly to their conventional counterparts when aug-cc-pVTZ or larger basis sets are used, in the absence of counterpoise correction the local approximation becomes significantly worse for the aug-cc-pVDZ basis set. The changes due to local correlation approximations for the aug-cc-pVDZ basis are reduced when diffuse functions are neglected for hydrogen atoms.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


