
Abstract
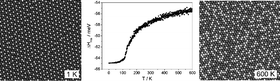
The energetics determining the distinct short-range order in two-dimensional (2D) monolayer CuxPd1−x surface alloys on a Ru(0001) substrate were investigated by Monte Carlo simulations and density functional theory calculations. Using a 2D lattice gas Hamiltonian based on effective pair interaction (EPI) parameters, the EPIs were derived for different Cu concentrations with Monte Carlo (MC) simulations by comparing with the atomic distributions obtained from atomic resolution
 Please wait while we load your content...
Please wait while we load your content...

