
Abstract
Ring closure reactions were investigated in a combined computational (density functional theory) and experimental study, to uncover the origin of diastereoselection in 5-exo-trig cyclizations of
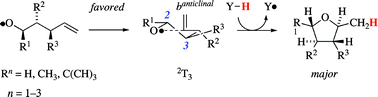
Theory states that the favored mode of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 entity to adopt positions that are associated with the fewest and least severe synclinal and synperiplanar interactions. A transition structure notation is proposed based on conformational characteristics of the heterocycle, the intermediates structurally resemble the closest, i.e.
CH2 entity to adopt positions that are associated with the fewest and least severe synclinal and synperiplanar interactions. A transition structure notation is proposed based on conformational characteristics of the heterocycle, the intermediates structurally resemble the closest, i.e.
The new transition state model serves as an alternative to cyclohexane-based guidelines and adequately addresses hitherto unsettled instances properly, such as the lack in diastereoselectivity observed in the 1-phenyl-4-penten-1-oxyl radical 5-exo-trig ring closure.
 Please wait while we load your content...
Please wait while we load your content...

