We present a parallel implementation of the multi-bank k·p code (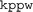 ) for calculation of the electronic structure and optical properties of zinc blend structure semiconductor quantum dots. The electronic wave-functions are expanded in a plane wave basis set in a similar way to ab initio calculations. This approach allows one to express the strain tensor components, the piezoelectric field and the arbitrary shape of the embedded quantum dot in the form of coefficients in the Fourier transform, significantly simplifying the implementation. Most of the strain elements can be given in an analytical form, while very complicated quantum dot shapes can be modelled as a linear combination of the Fourier transform of several characteristic shapes: box, cylinder, cone etc. We show that the parallel implementation of the code scales very well up to 512 processors, giving us the memory and processor power to either include more bands, as in the dilute nitrogen quantum dot structures, or to perform calculations on bigger quantum dots/supercells structures keeping the same “cut-off” energy. The program performance is demonstrated on the pyramidal shape InAs/GaAs, dilute nitrogen InGaAsN, and recently emerged volcano-like InAs/GaAs quantum dot systems.
) for calculation of the electronic structure and optical properties of zinc blend structure semiconductor quantum dots. The electronic wave-functions are expanded in a plane wave basis set in a similar way to ab initio calculations. This approach allows one to express the strain tensor components, the piezoelectric field and the arbitrary shape of the embedded quantum dot in the form of coefficients in the Fourier transform, significantly simplifying the implementation. Most of the strain elements can be given in an analytical form, while very complicated quantum dot shapes can be modelled as a linear combination of the Fourier transform of several characteristic shapes: box, cylinder, cone etc. We show that the parallel implementation of the code scales very well up to 512 processors, giving us the memory and processor power to either include more bands, as in the dilute nitrogen quantum dot structures, or to perform calculations on bigger quantum dots/supercells structures keeping the same “cut-off” energy. The program performance is demonstrated on the pyramidal shape InAs/GaAs, dilute nitrogen InGaAsN, and recently emerged volcano-like InAs/GaAs quantum dot systems.

You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
