
Insertion reactions of alkynes and organic isocyanides into the palladium–carbon bond of dimetallic Fe–Pd alkoxysilyl complexes†‡
Abstract
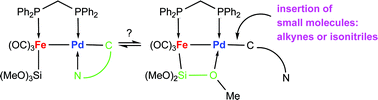
Insertion of MeO2C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CO2Me (DMAD) into the Pd–C bond of the heterodimetallic complex [(OC)3F
C–CO2Me (DMAD) into the Pd–C bond of the heterodimetallic complex [(OC)3F![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) e{μ-Si(OMe)2(
e{μ-Si(OMe)2(![[lower bond 1 start]](https://www.rsc.org/images/entities/char_e012.gif) OMe)}(μ-dppm)P
OMe)}(μ-dppm)P![[lower bond 1 end]](https://www.rsc.org/images/entities/char_e013.gif)
![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) d(dmba-C)] (2) (dppm = Ph2PCH2PPh2, dmba-C = metallated dimethylbenzylamine) and [(OC)3{(MeO)3Si}Fe(μ-dppm)Pd(8-mq-C,N)] (3) (8-mq-C,N = cyclometallated 8-methylquinoline) yielded the σ-alkenyl complexes [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(CO2Me)
d(dmba-C)] (2) (dppm = Ph2PCH2PPh2, dmba-C = metallated dimethylbenzylamine) and [(OC)3{(MeO)3Si}Fe(μ-dppm)Pd(8-mq-C,N)] (3) (8-mq-C,N = cyclometallated 8-methylquinoline) yielded the σ-alkenyl complexes [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(CO2Me)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CO2Me)(o-C6H4CH2NMe2)}] (7) and [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(CO2Me)C(CO2Me)(CH2C9H6N)}] (8), respectively. The latter afforded the adduct [(OC)3{(MeO)3Si}Fe(μ-dppm)Pd{C(CO2Me)C(CO2Me)(CH2C9H6N)}(CNBut)] (9) upon reaction with 1 equiv. of ButNC. The heterodinuclear σ-butadienyl complexes [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(Ph)C(Ph)C(CO2Me)(CO2Me)(o-C6H4CH2NMe2)}] (11) and [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(Ph)C(CO2Et)C(Ph)C(CO2Et)(CH2C9H6N)}] (13) have been obtained by reaction of the metallate K[Fe{Si(OMe)3}(CO)3(dppm-P)] (dppm = Ph2PCH2PPh2) with [PdCl{C(Ph)C(Ph)C(CO2Me)C(CO2Me)(o-C6H4CH2NMe2)}] or [PdCl{C(Ph)C(CO2Et)C(Ph)(CO2Et)}(CH2C9H6N)], respectively. Monoinsertion of various organic e(μ-dppm)Pd{C(NR)(CH2C9H6N)}] (15a R = Ph, 15b R = xylyl), a static six-membered C,N chelate is formed at the Pd centre, in contrast to the situation in [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(NR)(o-C6H4CH2NMe2)}] (14a R = o-anisyl, 14b R = 2,6-xylyl) where formation of a µ-η2-Si–O bridge is preferred over NMe2 coordination. The outcome of the reaction of the dimetallic e{μ-Si(OMe)2(OMe)}(μ-dppm)PdMe] with RNC depends both on the stoichiometry and the electronic donor properties of the e{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(N-o-anisyl)Me}] (16) results from the reaction in a 1 : 1 ratio. Addition of three equiv. of e{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{[C(N-o-anisyl)]3Me}] (18). After addition of a fourth equivalent of o-anisylNC, exclusive formation of the e(μ-dppm)Pd{[C(N-o-anisyl)]3Me}(CN-o-anisyl)] (19) was spectroscopically evidenced. In the complex [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{[C(N-o-C6H4COCH2)]2Me}] (20), the σ-bound diazabutadienyl unit is part of a 12-membered organic macrocyle which results from bis(insertion) of e{μ-Si(OMe)2(OMe)}(μ-dppm)PdMe]. In contrast, addition of two equivalents of e(μ-dppm)Pd{C(NBut)Me}(CNBut)] (21) in which both a terminal and an inserted
C(CO2Me)(o-C6H4CH2NMe2)}] (7) and [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(CO2Me)C(CO2Me)(CH2C9H6N)}] (8), respectively. The latter afforded the adduct [(OC)3{(MeO)3Si}Fe(μ-dppm)Pd{C(CO2Me)C(CO2Me)(CH2C9H6N)}(CNBut)] (9) upon reaction with 1 equiv. of ButNC. The heterodinuclear σ-butadienyl complexes [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(Ph)C(Ph)C(CO2Me)(CO2Me)(o-C6H4CH2NMe2)}] (11) and [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(Ph)C(CO2Et)C(Ph)C(CO2Et)(CH2C9H6N)}] (13) have been obtained by reaction of the metallate K[Fe{Si(OMe)3}(CO)3(dppm-P)] (dppm = Ph2PCH2PPh2) with [PdCl{C(Ph)C(Ph)C(CO2Me)C(CO2Me)(o-C6H4CH2NMe2)}] or [PdCl{C(Ph)C(CO2Et)C(Ph)(CO2Et)}(CH2C9H6N)], respectively. Monoinsertion of various organic e(μ-dppm)Pd{C(NR)(CH2C9H6N)}] (15a R = Ph, 15b R = xylyl), a static six-membered C,N chelate is formed at the Pd centre, in contrast to the situation in [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(NR)(o-C6H4CH2NMe2)}] (14a R = o-anisyl, 14b R = 2,6-xylyl) where formation of a µ-η2-Si–O bridge is preferred over NMe2 coordination. The outcome of the reaction of the dimetallic e{μ-Si(OMe)2(OMe)}(μ-dppm)PdMe] with RNC depends both on the stoichiometry and the electronic donor properties of the e{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{C(N-o-anisyl)Me}] (16) results from the reaction in a 1 : 1 ratio. Addition of three equiv. of e{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{[C(N-o-anisyl)]3Me}] (18). After addition of a fourth equivalent of o-anisylNC, exclusive formation of the e(μ-dppm)Pd{[C(N-o-anisyl)]3Me}(CN-o-anisyl)] (19) was spectroscopically evidenced. In the complex [(OC)3Fe{μ-Si(OMe)2(OMe)}(μ-dppm)Pd{[C(N-o-C6H4COCH2)]2Me}] (20), the σ-bound diazabutadienyl unit is part of a 12-membered organic macrocyle which results from bis(insertion) of e{μ-Si(OMe)2(OMe)}(μ-dppm)PdMe]. In contrast, addition of two equivalents of e(μ-dppm)Pd{C(NBut)Me}(CNBut)] (21) in which both a terminal and an inserted
 Please wait while we load your content...
Please wait while we load your content...

