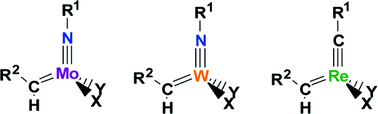
DFT(B3PW91) calculations have been carried out to rationalise the structural, electronic and spectroscopic properties of Mo and W imido M(NR1)(CHR2)(X)(Y) olefin metathesis catalysts by using either simplified or actual ligands of the experimental complexes. The calculated structures, energetics (preference for the syn isomer and alkylidene rotational barrier for the syn/anti interconversion), and spectroscopic properties (NMR JC–H coupling constants) are in good agreement with available experimental data. Additionally, the alkylidene νC–H stretching frequencies, not available experimentally, have been calculated. These quasi-tetrahedral complexes have a linear imido group and a C–H alkylidene agostic interaction, which stabilizes the syn isomer. Whether looking at M(NR1)(CHR2)(X)(Y), M = Mo, W, or the isolobal Re complexes, Re(CR1)(CHR2)(X)(Y), a linear correlation is obtained between both the alkylidene νC–H stretching frequencies and JC–H coupling constants with the calculated alkylidene C–H bond lengths. These correlations show that the strength of the α-C–H agostic interaction increases from alkylidyne Re to imido group 6 complexes and from Mo to W. The NBO and AIM Bader analyses show firstly that the imido and alkylidyne groups are both triply bonded to the metal, but that the triply bonded imido ligand is a weaker electron donor than the alkylidyne, hence the stronger α-C–H agostic interaction for group 6 imido complexes. Secondly, one of the π bonds of the triply bonded ligand is weakened at the transition state of the alkylidene rotation: while no lone pair is formed, the metal–ligand triple bond is polarized. This is more favourable for an imido than for an alkylidyne ligand, hence the lower alkylidene rotational barrier for the former complexes. Conversely, the aryl imido is even less of an electron donor than the alkyl imido group, which in turn strengthens the α-C–H agostic interaction and lowers the alkylidene rotational barrier even more.

 Please wait while we load your content...
Please wait while we load your content...

