
A time-dependent Hartree–Fock approach for studying the electronic optical response of molecules in intense fields
Abstract
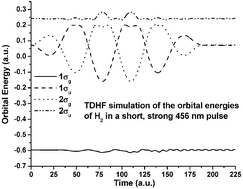
For molecules in high intensity oscillating electric fields, the time-dependent Hartree–Fock (TDHF) method is used to simulate the behavior of the electronic density prior to ionization. Since a perturbative approach is no longer valid at these intensities, the full TDHF equations are used to propagate the electronic density. A unitary transform approach is combined with the modified midpoint method to provide a stable and efficient algorithm to integrate these equations. The behavior of H2+ in an intense oscillating field computed using the TDHF method with a STO-3G basis set reproduces the analytic solution for the two-state coherent excitation model. For H2 with a 6-311++G(d,p) basis set, the TDHF results are nearly indistinguishable from calculations using the full time-dependent Schrödinger equation. In an oscillating field of 3.17 × 1013 W cm−2 and 456 nm, the molecular orbital energies, electron populations, and atomic charges of H2 follow the field adiabatically. As the field intensity is increased, the response becomes more complicated as a result of contributions from excited states. Simulations of N2 show even greater complexity, yet the average charge still follows the field adiabatically.
 Please wait while we load your content...
Please wait while we load your content...

