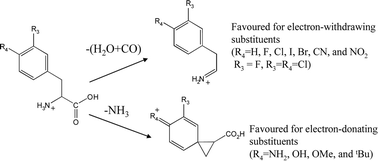
The gas-phase fragmentation of protonated phenylalanine and a series of its derivatives (tyrosine, 4-methylphenylalanine, 4-aminophenylalanine, 4-methoxyphenylalanine, 4-tert-butylphenylalanine, 4-fluorophenylalanine, 4-chlorophenylalanine, 4-bromophenylalanine, 4-iodophenylalanine, 4-cyanophenylalanine, 4-nitrophenylalanine, 3-fluorophenylalanine, and 3,4-dichlorophenylalanine) were examined using a combination of low energy CID in a quadrupole ion trap mass spectrometer as well as DFT calculations and RRKM modelling. In particular, the relationship between the electron-donating ability of the substituent and the competitive losses of H2O + CO and NH3 were explored through the application of the Hammett equation. It was found that electron-donating substituents promote the loss of NH3, while electron-withdrawing substituents suppress the loss of NH3 and favour the H2O + CO loss fragmentation channel instead. These observations are consistent with a neighbouring group pathway operating for the loss of NH3. Molecular orbital calculation (at the B3LYP/6-31+G(d,p) level of theory) were also performed for a range of derivatives to compare the relative transition state energy barriers for three competing mechanisms: (i) the combined loss of H2O + CO, which is triggered by an initial intramolecular proton transfer from the ammonium group to hydroxyl OH, followed by the combined loss of H2O and CO to form an immonium ion; (ii) loss of NH3via an aryl assisted neighbouring group pathway to yield a phenonium ion; (iii) loss of NH3via a 1,2-hydride migration process, which results in the formation of a benzyl cation. The relative energy barriers for H2O + CO loss remain nearly constant, while that for both NH3 pathways increase as the substituent moves from electron-donating to electron-withdrawing. The relative transition state energy for loss of NH3via the aryl assisted neighbouring group pathway is always lower than that of the 1,2-hydride migration process. RRKM modelling of the DFT predicted barrier heights suggest that the rate constants for H2O + CO loss are insensitive to the substituent on the ring, while the NH3 loss channels are greatly affected by the substituent. These theoretical results are consistent with the experimental observation of the relative yields of the competing fragmentation channels. Finally, comparisons with published gas phase and condensed phase studies on related systems are made.

 Please wait while we load your content...
Please wait while we load your content...

