
Accelerated, energy-conserving Born–Oppenheimer molecular dynamics via Fock matrix extrapolation
Abstract
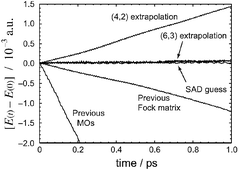
Born–Oppenheimer molecular dynamics calculations, especially those that exploit information retained from previous time steps in order to accelerate convergence of the electronic structure calculations, can suffer from systematic error in the energy gradient that manifests as a drift in the microcanonical energy. Here, we demonstrate that this is only the case when the self-consistent field (SCF) convergence criterion is set too low; using only a marginally tighter threshold (still two orders of magnitude lower than what is standard for geometry optimizations), the drift disappears completely, for a time scale of several picoseconds. Using a Fock matrix extrapolation technique, SCF convergence is achieved in as few as three iterations per time step, without sacrificing energy conservation. In test calculations for C2F4, (H2O)4−, (H2O)6, and [Fe(H2O)6]2+, we demonstrate energy-conserving Fock matrix extrapolation that reduces the number of SCF cycles by up to 70% and reduces the computer time per molecular dynamics step by 45–55%, relative to simulations performed without extrapolation.
 Please wait while we load your content...
Please wait while we load your content...

