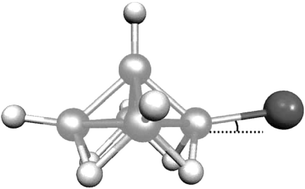
The molecular structures of 1-bromo–pentaborane(9) and 2-bromo–pentaborane(9) in the gas phase have been determined by electron diffraction and ab initio and DFT computational methods. Computational methods have also been applied to the fluoro and chloro analogues, to 1,2-dibromo-pentaborane(9), and to the parent unsubstituted borane. The electronic effects of halogen substitution on the borane cage are remarkably small, particularly for chlorine and bromine substituents, and steric effects are also minimal, even in the compound with two bromine atoms. The largest effects are (a) lengthening of B(base)–B(apex) bonds adjacent to the halogen in the 2-isomers, with an associated shortening of the opposite base–apex bond, (b) shortening of the B(base)–B(apex) bond in the 1-fluoro compound, and (c) increase of the B(base)–B(apex)–F angle in 1-F–B5H8, but a decrease in this angle in the 2-bromo compounds.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?