Computer simulation techniques have been used to investigate the defect chemistry of perovskite-structured ionic conductors based upon AZrO3
(A = Ca, Ba) and LaMO3
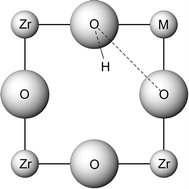
(M = Sc, Ga). Our studies have examined dopant site-selectivity, oxide ion migration and dopant–defect association at the atomic level. The energetics of dopant incorporation in AZrO3 show strong correlation with ion size. We predict Y3+ to be one of the most favourable dopants for BaZrO3 on energetic grounds, which accords with experimental work where this cation is the commonly used acceptor dopant for effective proton conduction. Binding energies for hydroxy–dopant pairs in BaZrO3 are predicted to be favourable with the magnitude of the association increasing along the series Y < Yb < In < Sc. This suggests that proton mobility would be very sensitive to the type of acceptor dopant ion particularly at higher dopant levels. Oxygen vacancy migration in LaScO3 is via a curved pathway around the edge of the ScO6 octahedron. Dopant–vacancy clusters comprised of divalent dopants (Sr, Ca) at the La site have significant binding energies in LaScO3, but very low energies in LaGaO3. This points to greater trapping of the oxygen vacancies in doped LaScO3, perhaps leading to higher activation energies at increasing dopant levels in accord with the available conductivity data.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


