
Substituent effects on the S–H bond dissociation energies of thiophenols†
Abstract
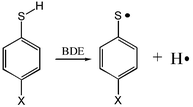
Density function UB3LYP/6-311++g(d,p) and perturbation theory ROMP2/6-311++g(d,p) calculations were performed on 4-substituted thiophenols and their corresponding radicals. It was found that although UB3LYP and ROMP2 methods underestimated the absolute S–H bond dissociation energies, they could predict almost as good relative S–H bond dissociation energies as a method of a considerably higher level, UCCSD(T)/6-311++g(d,p). From the calculation results it was determined that the S–H bond dissociation energies of thiophenols should have a positive correlation with the substituent σp+ constants whose slope was ca. 2.5 kcal mol−1. Such a slope indicated that the experimental S–H bond dissociation energies obtained from a previous solution phase measurement were reasonably accurate for para H, CH3, OCH3, Cl,
and NO2 substituted thiophenols. However, the solution phase bond dissociation energy for
 Please wait while we load your content...
Please wait while we load your content...