DOI:
10.1039/D6YA00006A
(Paper)
Energy Adv., 2026, Advance Article
Role of pH in tailoring Ni–Co hydroxide nanostructures for energy storage applications
Received
9th January 2026
, Accepted 1st May 2026
First published on 5th May 2026
Abstract
Nano-sized bimetallic nickel–cobalt hydroxide [NiCo(OH)4] with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Co/Ni atomic ratio was synthesized via a surfactant-free co-precipitation–hydrothermal method, where the solution pH was systematically varied to direct structural evolution. The growth process begins with the nucleation of hydroxide nanoplates, followed by dissolution and recrystallization into stacked, hexagonal, disc-like architectures. Among the prepared samples, the material synthesised at pH 9.0 exhibited the most favourable electrochemical behaviour, delivering a high specific capacitance of 621 F g−1 at 0.5 A g−1 and retaining 400 F g−1 at 10 A g−1, surpassing samples obtained at pH 8.0, 10.0, and 11.5. Morphological, structural, and spectroscopic characterisation confirmed that the enhanced performance of NCH-2 arises from its porous nanostructure, defect centres, and improved ion/electron transport kinetics for energy storage applications.
:1 Co/Ni atomic ratio was synthesized via a surfactant-free co-precipitation–hydrothermal method, where the solution pH was systematically varied to direct structural evolution. The growth process begins with the nucleation of hydroxide nanoplates, followed by dissolution and recrystallization into stacked, hexagonal, disc-like architectures. Among the prepared samples, the material synthesised at pH 9.0 exhibited the most favourable electrochemical behaviour, delivering a high specific capacitance of 621 F g−1 at 0.5 A g−1 and retaining 400 F g−1 at 10 A g−1, surpassing samples obtained at pH 8.0, 10.0, and 11.5. Morphological, structural, and spectroscopic characterisation confirmed that the enhanced performance of NCH-2 arises from its porous nanostructure, defect centres, and improved ion/electron transport kinetics for energy storage applications.
1. Introduction
Energy demand has increased rapidly in recent years. The need for alternative and green energy sources arises from the depletion of fossil fuel reserves and the resulting carbon emissions from their use.1,2 To address these issues and keep up with the always-rising need for energy, renewable energy sources, including wind, solar, and hydroelectricity, should be encouraged. Due to the intermittent nature of these renewable resources, efficient energy storage systems must be improved. Among various energy storage systems, electrochemical energy storage is most widely used due to its high efficiency, long cycle life, and design versatility. It mainly includes secondary ion batteries and supercapacitors, the latter comprising electric double-layer capacitors (EDLCs) and pseudo-capacitors (PCs). EDLCs store charge through ion adsorption at the electrode–electrolyte interface, offering excellent cycling stability but low capacitance (∼100 F g−1). PCs, in contrast, involve fast surface redox reactions, yielding 10–50 times higher capacitance but limited stability. Carbon-based materials are commonly employed for EDLCs, while transition metal oxides such as RuO2 and IrO2 serve as high-capacitance PC electrodes despite cost constraints. Therefore, the first-row transition metal layered hydroxides, particularly Co and Ni hydroxides, have been the subject of many studies.3,4 Better specific capacitance and rate capacity have been reported for hydroxides compared to oxides.5 Our group has reported significant oxygen vacancies in α-FeCo2(OH)6, which may act as functional centres for charge storage.6 In addition to the defects, crystallinity and crystal structure (space group-P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1),7 the morphology consisting of surface area to volume ratio,8 average pore size and size distribution,9 accessibility of redox-active sites, and mass transport channels10 affect the electrochemical performance of an electrode material. It has been reported that these parameters can be tuned by varying the synthesis parameters during hydrothermal synthesis of electrode materials. Among the synthesis parameters, reaction temperature, hydrothermal time, pH of the solution, and reaction chemistry can be varied to achieve suitable material parameters for the best electrochemical properties.11–13 Transition metal hydroxides such as NiCo(OH)4 (NCH) crystallise in α, β, and γ phases, where the β-phase (Pm1) is stable, the α-phase is metastable, and the γ-phase is unstable. The α-phase, with a larger interlayer spacing (∼7.8 Å), facilitates ion intercalation/deintercalation, enhancing capacitance. During cycling, α → β transformation occurs, while overcharging may reverse it. Co(OH)2 exhibits a high theoretical capacitance (∼3600 F g−1). In bimetallic hydroxides, synergistic interactions between metal ions enhance electrical conductivity, ion diffusion, and active site density. For example, FeCo2(OH)6 with oxygen vacancies on Co sites shows improved redox activity, demonstrating that bimetallic synergy significantly boosts electrochemical performance.
m1),7 the morphology consisting of surface area to volume ratio,8 average pore size and size distribution,9 accessibility of redox-active sites, and mass transport channels10 affect the electrochemical performance of an electrode material. It has been reported that these parameters can be tuned by varying the synthesis parameters during hydrothermal synthesis of electrode materials. Among the synthesis parameters, reaction temperature, hydrothermal time, pH of the solution, and reaction chemistry can be varied to achieve suitable material parameters for the best electrochemical properties.11–13 Transition metal hydroxides such as NiCo(OH)4 (NCH) crystallise in α, β, and γ phases, where the β-phase (Pm1) is stable, the α-phase is metastable, and the γ-phase is unstable. The α-phase, with a larger interlayer spacing (∼7.8 Å), facilitates ion intercalation/deintercalation, enhancing capacitance. During cycling, α → β transformation occurs, while overcharging may reverse it. Co(OH)2 exhibits a high theoretical capacitance (∼3600 F g−1). In bimetallic hydroxides, synergistic interactions between metal ions enhance electrical conductivity, ion diffusion, and active site density. For example, FeCo2(OH)6 with oxygen vacancies on Co sites shows improved redox activity, demonstrating that bimetallic synergy significantly boosts electrochemical performance.
It is well reported in the literature that α-TM hydroxide samples exhibit better initial energy density but demonstrate poorer cycling stability and rate capability than β-TM hydroxides [Yin et al.,14 Akhtar et al.15 and Rajkamal et al.16]. Yi et al.17 have reported that α/β phase synergies lead to improved electrochemical performance. Optimized α/β mixed-phase samples show enhanced capacity and superior capacity retention (∼108% after 1000 cycles at a current density of 20 mA cm−2) compared to ∼ 50% in the α-phase and ∼87% in the β-phase. The improved performance is attributed to the more stable structure and better electrical conductivity of the β-phase, along with the higher interlayer spacing in the α-phase, which facilitates efficient ion intercalation and charge transport. These combined effects are likely responsible for the superior electrochemical performance observed in the α/β mixed-phase sample in this study. Akhtar et al. have synthesised nanoflowers of Ni(OH)2 and have achieved a high capacitance of 1511 F g−1 at a current density of 10 mA cm−2 and 100% capacity retention after 5000 cycles at 50 mA cm−2. The cycling stability is higher for the mixed phase than both α and β pure phases. These studies propose a synergy between better phase stability of the β phase and higher initial capacitance of the α phase; thus, an optimised ratio results in good initial capacitance and higher cycling as well as rate efficiencies, as observed in this study.
In the current study, NiCo(OH)4 (NCH) samples prepared under systematically varied pH conditions demonstrated marked differences in specific capacitance, highlighting the crucial role of synthetic control in achieving high-performance electrode materials. This approach underscores the importance of defect engineering and tailored nanostructures in advancing bimetallic hydroxide-based energy storage technologies. The pH of the reaction medium during synthesis plays a crucial role in influencing the morphology, defect concentration, structural stability, and electrical conductivity of the resulting materials. These factors directly affect the electrochemical performance, including specific capacitance, cycling stability, and rate capability.
Recent studies have highlighted that optimizing the pH conditions can significantly enhance electrochemical properties. For instance, Bhatt et al.18 reported that controlled pH conditions lead to improved specific capacitance (∼3061 F g−1 at 0.5 A g−1), excellent cycling stability (∼99% retention after 5000 cycles), and superior rate capability in Ni–Co-based hydroxide systems. Similarly, Wang et al.19 demonstrated that pH tuning influences the preferential growth direction and morphology of Ni–Co–Mn hydroxide nanostructures, which in turn enhances electrochemical performance. These studies clearly indicate that careful control of synthesis parameters such as pH is essential for optimizing electrochemical behavior. Hu et al.20 synthesized layered double Ni–Co hydroxide nanosheets by optimizing the pH conditions. The optimized material exhibited excellent electrochemical performance, delivering a high energy density of ∼48.57 Wh kg−1 at a power density of 800 W kg−1, along with nearly 100% capacity retention after 10000 cycles at a current density of 10 A g−1. Xiang-Yu You et al.21 reported the synthesis of transition metal hydroxides using a metal tetrafluoroborate precursor. In their study, the pH of the reaction medium was carefully controlled by varying the concentration of 2-methylimidazole, which effectively induced precipitation and influenced the structural evolution of the material. These studies further emphasize that controlled pH conditions during synthesis play a crucial role in tailoring the morphology and structural properties, and ultimately enhancing the electrochemical performance.
We establish a correlation between pH-dependent structural features and electrochemical performance. By combining structural characterization with detailed kinetic analysis using Dunn's model, this study reveals how an optimized balance between diffusion-controlled and capacitive contributions governs the superior performance of the NCH (pH-09) sample. This work provides deeper insight into the role of pH in tuning ion transport, defect states, and charge storage mechanisms, which is not extensively addressed in previous reports.
NCH nanostructures with varying proportions of α and β phases were synthesised hydrothermally using ammonia hydroxide to adjust the solution pH to 8.0, 9.0, 10.0, and 11.5, resulting in samples labelled NCH-1, NCH-2, NCH-3 and NCH-4, respectively. The sample formed at pH 9.0 (NCH-2) displayed the most favourable electrochemical properties, achieving a specific capacitance of 1230 F g−1 at 5 mV s−1, which was significantly higher than the other samples. Galvanostatic charge–discharge (GCD) measurements further confirmed its superior rate capability, delivering 622 F g−1 at 0.5 A g−1 and 400 F g−1 at 10 A g−1. Detailed characterisation demonstrated that its enhanced performance is due to a higher density of structural disorder/defect sites, such as cobalt vacancies and oxygen interstitials, which facilitate efficient charge storage and ideal ion diffusion. The enhanced electrochemical kinetics of the NCH series are consistent with recent advancements in defect engineering for transition metal hydroxides. Recent reports by Gao et al.22 highlight that the introduction of structural defects effectively tunes the electronic structure and enhances carrier concentration, thereby providing additional extrinsic pseudo capacitance. Furthermore, Bhatt et al.23 have identified that oxygen vacancies and lattice distortions in nickel–cobalt nanostructures act as catalytic centres that lower charge transfer resistance and optimize ion adsorption. Consequently, the structural disorder inferred from our XRD and electrochemical data suggests a high density of active sites, facilitating the superior rate performance of the NCH-2 electrode. A comparison of the electrochemical performance of the sample NCH-2 with other bimetallic hydroxide and oxide materials without complex compositional modifications reported in the literature is shown in Table S1.
2. Experimental section
2.1. Materials
Cobalt (II) nitrate hexahydrate (Co(NO3)2·6H2O) and nickel nitrate hexahydrate (Ni(NO3)2·6H2O) were procured from LOBA CHEMIE Pvt. Ltd. India. Ammonium hydroxide (NH4OH, 25% NH3) and potassium hydroxide (KOH) were obtained from Sigma-Aldrich (Lyon, France), and Emplura/Merck Life Science Pvt. Ltd, respectively. Chemical reagents are of analytical grade and were used as received without further purification. Double distilled water was used throughout the experiments.
2.2. Synthesis process
A Co-precipitation/hydrothermal method was used to synthesized nano-sized bimetallic NiCo(OH)4 (NCH) using ammonia hydroxide (NH4OH, 25%) to adjust the solution pH to 8.0, 9.0, 10.0, and 11.5, resulting in samples labelled NCH-1, NCH-2, NCH-3 and NCH-4 respectively. A standard process involved dissolving 1.48 g of (nickel and cobalt (1:1)) nitrate hexahydrate in 60 mL of deionised (DI) water. The respective amount of NH4OH was added dropwise into the above solution and to ensure complete mixing, the solution was then magnetically stirred for 1 hour. The resultant suspension was put into a stainless-steel autoclave lined with Teflon and heated for 4 hours at 120 °C. To get the final powder samples, the precipitate was collected after naturally cooling to room temperature, extensively cleaned many times with ethanol and DI water to get rid of any remaining ions and contaminants, and then dried at 70 °C overnight.
2.3. Characterization
Synchrotron X-ray diffraction (SXRD) was used to study the crystallographic phases, particle size, and microstructure in the synthesised powder samples. At Indian synchrotron Indus-2, the synchrotron measurements were performed on an angle dispersive X-ray diffraction beamline (BL-12) with an X-ray wavelength of 0.719 Å.24 The morphology and the composition were analysed by FESEM and EDX, respectively (Model: JSM-7610F). FESEM images were captured for the powder samples. Furthermore, the oxidation states were confirmed by XPS analysis (Model: Thermo Fisher Scientific Escalab Xi+). Electrochemical measurements (CV, GCD, and EIS) were conducted using platinum mesh as the counter electrode and a saturated calomel electrode as the reference electrode in a Biologic SAS, SP-150e (3-electrode Workstation) and CR 2032 coin cell measurements were done using a Bio-logic SAS Battery health cycler system at Battery Fabrication Lab, UPES Dehradun.
3. Results and discussion
3.1. X-ray diffraction patterns
Fig. 1 presents the XRD patterns of the Ni–Co hydroxide (NCH) samples synthesised at varying pH levels. Both Ni(OH)2 and Co(OH)2 exhibit two hexagonal polymorphs, α and β, differing in their layer stacking and interlayer composition. In the α-phase, a hydroxyl-deficient phase characterized by the absence of OH− anions and the presence of interlayer anions (NO3−), the layers can be stacked randomly along the c-axis with water or anionic intercalates residing within the van der Waals gap. On the other hand, they can be well-ordered and closely packed along the c-axis, as in the β-phase, which has a stoichiometric composition and an interlayer separation of 4.6 Å. The α-phase can form when ammonia is used as the precipitation agent. This is because it can insert anions (NO3−) and small molecules (NH3, H2O) into the structure between the layers. After the hydrothermal process, the stoichiometric β-phase is the more stable form because of the process of dissolving and recrystallising25 and its compact brucite-like structure, higher crystallinity, and minimized surface energy under prolonged alkaline hydrothermal conditions. This process enhances phase purity and structural integrity, yielding β-NiCo(OH)4 with superior cycling stability and conductivity. The diffraction patterns of the four samples show peaks at 2θ = 8.98°, 15.23°, 17.74°, 23.67°, 26.55°, and 28.07°, with d-spacings of 4.63, 2.74, 2.35, 1.77, 1.58, and 1.5 Å, respectively. These are the (001), (100), (002), (021), (003), and (111) planes, in that order, of the hexagonal structure and can be precisely linked to the β phase26,27 (JCPDS 1074–1057). The lattice parameters estimated are a = b = 3.1639 Å; c = 4.6343 Å; α = β = 900; γ = 1200 and are in good agreement with the literature. Also, the peaks for ±(001), ±(002), ±(110), and ±(101) can be seen at about 5.30°, 10.72°, 16.12°, and 29.24°, with d-spacings of 7.85, 3.88, 2.59, and 1.44 Å. The α-phase is also represented by the same space group as that of the β-phase, but with a larger c parameter. The lattice parameters for this phase are a = b = 3.1682 Å; c = 7.8469 Å, which are in good agreement with the literature.28 As seen from Fig. 1, all four samples contain a mixture of the two phases. Since the space group of both phases is the same, we estimate the molar concentration of the phases by comparing the area under the curve for the same Bragg reflection (reflection (001) in this case). The molar concentration of the α (β) phase in all samples is mentioned in Table S2. The concentrations of α (β) phases were approximately estimated by taking the ratio of the area under the highest intensity XRD peaks normalised by (A/|Fhkl|)2 in the two phases, where Fhkl is the unit cell scattering factor for diffraction by (hkl) planes and A is the sum of the atomic numbers of all the atoms in the unit cell. The difference in geometric factors like Lorentz polarisation factor and the absorption factor has been neglected in this estimation of phase ratios. We observe that the sample having lower (pH-09) NCH-2 has the lowest concentration of the α-phase, followed by the samples NCH-3, NCH-1, and NCH-4. We have estimated the particle size of samples and micro-strain using the Williamson–Hall (W–H) relation.29| | |
βcosθ = 4·εsinθ + Kλ/d
| (1) |
 |
| | Fig. 1 (a) The XRD of NCH samples, and (b)–(d) the W–H plots for NCH-1, NCH-2, and NCH-3 samples. | |
In eqn (1), β, ε, λ, K, and d are the full width at half maxima (FWHM) of the XRD Bragg peak at diffraction peak 2θ, micro strain, wavelength of the X-rays, a constant (K = 0.9), and particle size, respectively. We have estimated the micro-strain along (00h) by using (001), (002), (003), and (004) diffraction peaks of the β-phase. The W–H plots for the three samples, NCH-1, NCH-2, and NCH-3, are shown in Fig. 1(b–d). For sample NCH-4, the W–H plot could not be achieved because of the small peak intensities of the β-phase. The particle size and the micro-strain were calculated from the intercept and the slope of the linear fit, as shown in Fig. 1(b–d). The particle size and micro-strain are summarised in Table S2. It may be noted that the particle size of the β-phase for the sample NCH-4 and that of the α-phase for all the samples are estimated using the Scherrer formula.30 The α-phase of all the samples shows lower-sized particles (10–16 nm) compared to the β-phase (32–45 nm). The micro-strain of samples NCH-2 and NCH-3 is found to be much lower (0.06 ± 0.01%) than that of NCH-1 (0.145%).
3.2. Fourier transform infrared (FTIR) spectroscopy
The FTIR spectra of the four samples are shown in Fig. S1. The spectra of all the samples show similar absorption bands. A broad and strong absorption band at 3621 cm−1 is related to O–H stretching modes. A sharper peak for the sample NCH-4 suggested a well-ordered hydroxyl group in a well-crystallised structure, whereas the sample NCH-1 indicated more hydrogen bonding and interlayer water molecules. The spectra also showed bands at 2414 cm−1 and 2847 cm−1, which were related to C–O/C–H group antisymmetric and symmetric stretching modes, respectively. The stretching vibrations of free water molecules in the material are responsible for the peak at roughly 1645 cm−1. Additionally, the N–O band of NO−3, which is present in the material's interlayers, is responsible for the peak at 1384 cm−1. In NCH samples, Co/Ni–OH stretching vibrations are represented by the band at 768 cm−1. The stretching vibration of the Ni/Co–O bond is responsible for the peak at roughly 540 cm−1. (NCH 2 and NCH-3 samples show the sharpest Ni/Co–O bond, suggesting a better crystalline nature).31
3.3. Morphology and surface area studies
3.3.1. Field emission scanning electron microscopy and EDX. The surface morphology of the four samples in the form of electrodes (active material 80%, carbon black 10%, PVDF 10%) is examined by micro-graphing the surface using FESEM. The high magnification (8 ± 1k) (scale bar: 1 µm) images are shown in Fig. 2(a–d). The overall morphology in all the samples seems to consist of disc-like structures, with a diameter of 1 to 2 µm. However, in samples NCH-1 and NCH-4, the morphology consists of micron-sized discs overlayed on finer particles. In general, the particles in all the samples are big, densely packed, and have slight asymmetry (disc-like). The FE-SEM image of sample NCH-2 at a resolution of 9 K shows more uniform and well-defined discs, and the morphology's apparent openness is a sign of better ion transport channels. At 9 K resolution, the NCH-4 sample shows signs of losing its porosity and having structures that are highly clumped together and not well-defined.
 |
| | Fig. 2 FESEM images of NCH samples showing morphological variations: (a) NCH-1, (b) NCH-2, (c) NCH-3, and (d) NCH-4. Scale bars: 1 µm. | |
Elemental compositions in the four samples were studied using EDX analysis, as shown in Fig. S2. From EDX analysis, we observe that as the pH of the reactants during sample synthesis increases, the atomic percentage of Ni decreases, and that of Co increases in the NCH samples. The variation in the Ni/Co ratio is attributed to the pH-dependent precipitation behavior of the metal ions, which influences the structural and electrochemical properties of the samples.
3.3.2. Brunauer–Emmett–Teller (BET). BET characterises the specific surface area and the pore size distribution of the NCH samples. The adsorption–desorption isotherms of the samples are shown in Fig. S3. All the isotherms with convex shapes and low hysteresis area are of type V, which represents a mesoporous structure with small adsorbate–adsorbate interaction. The pore volume, BET surface area, and BJH surface area were estimated using a literature report by Barrett et al.32 and are summarised in Table S3. Sample NCH-2 shows a minimum BET (BJH) surface area of 12.84 m2 g−1 (13.86 m2 g−1), among the four samples reported in this paper. The same sample also shows the minimum pore volume. This is also corroborated by the FESEM images, where the sample NCH-2 seems most compact and dense. For moderate to high pH values of the reactant during the synthesis of NCH samples, the surface area increases with increasing pH value. The sample NCH-4, synthesised at a pH-11.5 value, showed maximum BET surface area (pore volume) of 52.33 m2 g−1 (0.298 cm3 g−1). A low pH environment inhibits controlled nucleation, promoting rapid crystal growth and aggregation, resulting in dense morphologies with decreased porosity. The limited availability of hydroxide ions restricts precursor hydrolysis and delays nucleation, resulting in non-uniform particle growth and poorly defined structures.In general, it is believed that a larger surface area presents higher storage capacity mainly through intercalation. However, due to various reasons like smaller electrical conductivity in porous systems,33 ionophobic channels, mostly in the case of MOFs and other porous materials,34 the electrochemical performance of samples is poorer.35 Additionally, despite having the lowest BET surface area, NCH-2 exhibits superior performance, indicating that pore structure and ion transport kinetics play a more decisive role than surface area alone.
3.4. X-ray photoelectron spectroscopy (XPS)
XPS was performed to examine the surface chemical composition and oxidation states of the NCH-1, NCH-2, NCH-3, and NCH-4 samples. The survey spectra confirm the presence of Ni, Co, and O elements in all samples (Fig. 3(a) for NCH-2 and Fig. S4 for NCH-1,3,4). For the NCH-2 sample, the Ni 2p3/2 region (Fig. 3b) exhibits distinct peaks at 854.60 eV (Ni2+), 855.89 eV (Ni3+), and a satellite peak at 860.59 eV. The Ni 2p1/2 region contains peaks at 872.09 eV (Ni2+), 873.42 eV (Ni3+), and a satellite at 878.66 eV. The presence of Ni3+ on the surface suggests the existence of defect states such as Ni vacancies or oxygen interstitials, which are known to improve electrochemical performance by serving as active centres for charge storage.36–39 The Co 2p3/2 region (Fig. 3c) exhibits peaks at 779.53 eV (Co3+), 781.13 eV (Co2+), and 784.68 eV (Co2+), while the Co 2p1/2 region reveals peaks at 794.67 eV (Co2+) and 796.04 eV (Co3+).40–42 The O-1s spectrum (Fig. 3d) shows two peaks at 529.17 eV and 530.12 eV, corresponding to lattice oxygen and metal–oxygen bonding, respectively.43,44 For the NCH-1 sample (Fig. S5(a–c)), the Ni 2p3/2 peak appears at 854.42 eV (Ni2+) and the Ni 2p1/2 peak at 872.04 eV (Ni2+), with no noticeable contributions from Ni3+. The Co 2p3/2 and 2p1/2 peaks are located at 780.40 eV and 796.23 eV, respectively, corresponding to Co2+. The O 1s spectrum displays peaks at 529.93 eV (O2−) and 531.26 eV (OH− species). In the NCH-3 sample (Fig. S5(d–f)), the Ni 2p3/2 spectrum reveals peaks at 854.55 eV (Ni3+) and 856.74 eV (Ni2+), while Ni 2p1/2 peaks are seen at 871.92 eV (Ni2+) and 873.53 eV (Ni3+).45,46 The Co 2p3/2 region shows peaks at 779.37 eV and 780.86 eV, both associated with Co2+, whereas the Co 2p1/2 region presents features at 795.06 eV (Co2+) and 796.51 eV (Co3+).47 The O-1s spectrum includes peaks at 528.76 eV (lattice oxygen) and 529.96 eV (metal–oxygen bonds). Similar to NCH-2, the presence of both Ni3+ and Co3+ indicates the existence of beneficial defect states. For the NCH-4 sample (Fig. S5(g–i)), Ni 2p3/2 peaks are observed at 854.76 eV (Ni2+) and 856.11 eV (Ni3+), while Ni 2p1/2 peaks appear at 872.36 eV (Ni3+) and 874.11 eV (Ni2+). The Co 2p3/2 spectrum shows peaks at 779.48 eV and 782.84 eV, both corresponding to Co3+, and the Co 2p1/2 region reveals peaks at 794.60 eV (Co3+) and 796.10 eV (Co2+). The O 1s region shows binding energies at 529.27 eV (metal oxides) and 530.16 eV (M–O–M bonding, where M = Ni or Co). Interestingly, the samples NCH-2 and NCH-3, which contain the majority β-phase composition, exhibit clear Co3+ peaks in their XPS spectra. These samples also display enhanced electrochemical performance compared to NCH-1 and NCH-4, which either lack Co3+ peaks or exhibit them in lower intensities. This correlation suggests that the presence of Co3+, indicative of Co vacancies or oxygen interstitials, acts as an effective functional site for charge storage, contributing to the improved electrochemical characteristics observed in NCH-2 and NCH-3.
 |
| | Fig. 3 XPS spectrum of NCH-2, (a) survey scan, and (b)–(d) Ni-2p, Co-2p, and O-1s edges, respectively. | |
3.5. Electrochemical results
The electrochemical studies of NCH-1, NCH-2, NCH-3, and NCH-4 electrodes having mass loadings of ∼3.7, 3.5, 3.8, and 3.9 mg cm−2, respectively, on a 1 cm2 Ni foam substrate were systematically evaluated in aqueous 3 M KOH electrolyte by using a 3-electrode electrochemical workstation.
3.5.1. Cyclic voltammetry (CV). Fig. 4(a) shows anodic and cathodic current as a function of applied potential at 10 mV s−1 scan rate. For the full CV graph from 1 to 100 mV s−1 see Fig. S6. The curves are used to initially assess the electrochemical performances of the electrodes in a 3-electrode configuration. A pair of redox peaks can be seen in their CV curves for low scan rates in the anodic peaks as well as for all the scan rates for cathodic peaks, which stem from the reversible reactions between Ni2+/Ni3+ and Co2+/Co3+, which are illustrated in eqn (1)–(4) in the electrochemical redox processes.48,49| | |
Ni(OH)2 + OH ⇌ NiOOH + H2O + e−
| (2) |
| | |
NiOOH + e− ⇌ NiO + OH
| (3) |
| | |
Co(OH)2 + OH ⇌ CoOOH + H2O + e−
| (4) |
| | |
CoOOH + OH ⇌ CoO2 + H2O + e−
| (5) |
 |
| | Fig. 4 (a)–(d) Cyclic voltammetry plot of NCH samples at 10 mV s−1. (b) Log–Log plot of specific capacitance vs. scan rate of NCH samples, (c) specific capacitance vs. scan rate of NCH samples and (d) GCD plot of NCH samples at 5 A g−1. | |
The power-law given in eqn (6) was used to study the charge storage mechanism in the electrodes. In Fig. 4(b), we plot the log (Ip) versus log(v) (see eqn (7)).50,51
where,
a and
b are Constant parameters; the value of parameter
b can be determined from a slope of the linear plot between log
Ip vs. log ν. The value of parameter
b depends on the charge storage mechanism. The value of
b is close to a value of 0.5 for charge storage by the diffusion-controlled intercalation process (DIP), whereas for surface-controlled capacitive processes (SCP) of charge storage, the value of
b is close to 1.
| | |
log(Ip) = log(a) + b·log(v)
| (7) |
| |
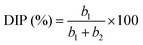 | (9) |
| |
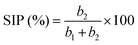 | (10) |
Eqn (8) can be rearranged as:
| |
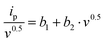 | (8*) |
A plot of (Ip/v0.5) versus (v0.5) yields a straight line, where (b1) and (b2) are obtained from the intercept and slope, respectively.
Dunn's analysis reveals that charge storage arises from both capacitive and diffusion-controlled processes, as indicated by b-values of 0.48–0.64 as shown in Table S4 and Fig. 4(b). Relative percentage contributions of DIP and SCP were quantified using eqn (8)–(10) and the NCH-2 and NCH-3 samples show a balanced contribution from both mechanisms, enabling efficient ion transport and improved electrochemical performance. In contrast, the higher diffusion contribution in NCH-4 may limit its rate capability. The specific capacitance as a function of scan rate was calculated using the integrated area under the CV curve and using eqn (11) given below:16
| |
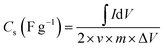 | (11) |
The specific capacitance as a function of the scan rate is plotted in Fig. 4(c). This illustrates the connection between specific capacitance and scan rate (which ranges from 1 to 100 mV s−1). It was found that at slower scan rates, there is more time for electrolyte ions to reach and interact with the electrode surface, resulting in a higher specific capacitance for the supercapacitor.52 The capacitance did, however, decrease somewhat when the scan rate increased from 50 to 100 mV s−1. This finding suggests that faster scan speeds restrict ions to the electrode surface, giving them less time to diffuse into the electrode's pores, and the trend is in agreement with literature.53 Out of the four samples, the NCH-2 sample shows much better specific capacitance. Typically, the specific capacitance at a scan rate of 5 mV s−1 was 1230 F g−1 observed for sample NCH-2 compared to 386 F g−1 for sample NCH-1 and 380 F g−1 for sample NCH-4. Sample NCH-3 shows a moderate specific capacitance of ∼880 F g−1.
3.5.2. Galvanostatic charge and discharge (GCD). The GCD plot of NCH samples at 5 A g−1 is shown in Fig. 4(d). The electrochemical performance of all the samples at various current densities was further evaluated, as shown in Fig. S7. The non-linear charging and discharging behaviour of these plots confirms that all four samples possess characteristics similar to those of battery-like behaviour.54,55 The specific capacitance of electrodes was calculated using eqn (12), and the values are listed in Table S5. Specifically, the samples NCH-2 and NCH-3 show better storage capacity than samples NCH-1 and NCH-4, in agreement with the CV results. As the current density increases, the number of active sites that can be reached decreases because ions have less time to move into the porous structure of the electrode material. NCH-2 has the best electrochemical performance among all the samples. It has the highest specific capacitance of 621 F g−1 at a current density of 0.5 A g−1, compared to 343 F g−1 at a discharge rate of 0.5 A g−1 for the sample NCH-4. We find a decrease in capacity as the discharge current density increases, in agreement with the literature.56 The rate capability of the samples was evaluated by comparing capacitance retention at higher current densities. The NCH-1, NCH-2, NCH-3, and NCH-4 samples retain approximately 42%, 75%, 65%, and 26% of their initial capacitance at 5 A g−1, respectively. Among these, NCH-2 and NCH-3 demonstrate superior electrochemical performance, exhibiting both higher specific capacitance and better rate capability.| |
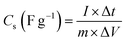 | (12) |
where I – applied current (mA), Δt – discharge time, and ΔV – potential window.The superior rate performance of sample NCH-2 compared to the other samples is because supercapacitor electrodes with low specific surface area (SSA) typically outperform high-SSA versions in rate capability thanks to their well-designed mesopores, which promote swift ion movement and decrease internal resistance, sidestepping the ion entrapment and extended pathways typical of the winding micropores in high-SSA materials. Consequently, they achieve stronger capacitance retention under high current loads, since overly high SSA elevates polarization and series resistance amid rapid cycling. Additionally, we have studied the surface morphology to comment on the reach of the electrolyte to the electrode active material. We further analyse the XRD data to find out the particle size, micro-strain, and fraction of the two stable phases (α and β), to see if the electrochemical performance has any correlation to these parameters. We have also studied the transition metal charge states and possible surface defect states using XPS measurements. It may be mentioned that the structural disordered/defect sites have been related to the centres promoting redox reactions.56
3.5.3. Electrochemical impedance spectroscopy (EIS). EIS is extensively used to probe the solution, as well as charge transfer resistance, and to assess electrode kinetics. Prior to EIS measurements, all electrodes were fully discharged to the lower potential limit and stabilized to ensure identical electrochemical states. The EIS data from NCH samples plotted in a Nyquist plot across the frequency range of 100 kHz to 10 mHz are shown in Fig. 5. The impedance spectra were fitted using an equivalent circuit of Rs − (Rct\CPE) − ZW. Here, Rs denotes the solution (electrolyte) resistance, and Rct signifies charge transfer (polarisation) resistance, which arises from the polarisation of the electrode, causing current flow through the electrochemical reaction occurring at the electrode surface. ZW represents the Warburg element, which is the impedance arising from changes in the diffusion layer and the rate of diffusion in the working electrode. CPE stands for the constant phase element, representing electric double-layer capacitance that does not exhibit ideal behaviour. This model was selected based on the characteristic Nyquist plot features, including a depressed semicircle at high frequency and a diffusion-controlled region at low frequency. The CPE impedance is expressed as57 ZCPE = 1/T·(jw)p, where T is the capacitance pre-factor and P is the frequency exponent. P values of 0 and 1 correspond to pure resistance and dielectric capacitance, respectively. A p-value between 0.8 and 1 indicates a non-ideal double-layer capacitance, with non-ideality arising from roughness, inhomogeneity, and mixed charge storage. We fitted the EIS data for four NCH samples, as summarised in Table S6. We observed that Rs (=0.21 Ω) and Rct (=0.38 Ω) for sample NCH-2 are the lowest among the four samples reported here. Rct (=0.52 Ω) is also low for NCH-3. This correlates well with the electrochemical performance of the samples, where NCH-2 and NCH-3 demonstrate better performance. The Warburg exponent ZW-P (∼0.5) for NCH-2 indicates ideal semi-infinite diffusion, while p > 0.5 for all other samples signifies finite length diffusion and therefore inferior electrochemical performance.58 Similarly, a conclusion was derived from the CPE exponent. For sample NCH-1, a low p (=0.41) indicates a poor-quality surface with a minimal form of kinetic reaction. NCH-3 and 4 show p ∼ 0.97, which is close to ideal capacitance. For sample NCH-2, p (=0.72) indicates double-layer capacitance behaviour in this frequency region. Thus, we conclude from the EIS analysis that sample NCH-2 exhibits ideal semi-infinite diffusion in the low frequency (Warburg) region and functions as double-layer capacitance in the high frequency region. Furthermore, the low chi square error value (χ2 ∼ 4.2 × 10−4) confirms the reliability of the fitting. These results are consistent with the superior electrochemical performance observed for the NCH-2 sample.
 |
| | Fig. 5 Nyquist plot of NCH samples (inset: equivalent electrical circuit). | |
4. Fabrication of a symmetric coin cell-2032 device
To evaluate practical applicability, we fabricated a pair of uniform NCH-2 electrodes, each featuring a 1 cm2 active surface and a total mass loading of ∼1.7 mg. To prevent short-circuiting, we employed a cellulose-based separator that had undergone a 24-hr saturation in the same alkaline 3 M KOH electrolyte. Finally, the device was assembled by placing the saturated separator between the electrodes and crimped tightly.59| |
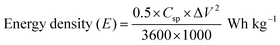 | (13) |
| |
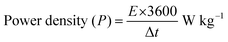 | (14) |
where Csp – specific capacitance, ΔV – potential window, and Δt – discharge time.
Cyclic voltammetry (CV) measurements were performed between 5 and 100 mV s−1, and displayed quasi-rectangular profiles with noticeable redox features, confirming the coexistence of an electric double-layer and faradaic charge storage as shown in Fig. S8(a). From Fig. S8(b and c), the steady rise in current response with increasing scan rate indicates favorable rate performance. The extracted b-values (∼ 0.54–0.56) suggest that charge storage is governed by a combination of surface-controlled and diffusion-limited processes, which is further supported by Dunn's analysis showing higher capacitive contribution at elevated scan rates. Galvanostatic charge–discharge (GCD) curves shown in Fig. S8(d) recorded within a 1.5 V window exhibited nearly symmetric triangular shapes, reflecting good reversibility. The specific capacitance (Table S7) decreased from 22.0 to 10.0 F g−1 as the current density increased from 0.25 to 1.0 A g−1, mainly due to kinetic limitations. The corresponding energy and power density were calculated from Equations (13) and (14) and the energy densities ranged from 6.86 to 3.13 Wh kg−1, while the power density increased from 178 to 714 W kg−1, demonstrating the expected trade-off between energy and power. The device retained ∼74% of its initial capacitance after 500 cycles with a Coulombic efficiency of ∼93%, indicating stable cycling behavior. Although the energy density is moderate, the Ragone plot (Fig. S9) demonstrates the practical applicability and stable performance of the symmetric device. Further optimization of electrode architecture and device configuration is expected to enhance the energy density.
Overall, the results confirm that the NCH-2 system exhibits reliable electrochemical performance with mixed charge storage characteristics suitable for practical applications.
5. Conclusions
This study demonstrates the successful synthesis of nano-sized bimetallic NiCo(OH)4 using a surfactant-free co-precipitation/hydrothermal approach, where pH was used as a key parameter to tune the structural and electrochemical properties. The sample prepared at pH 9 (NCH-2) exhibited the most favourable characteristics, achieving a high specific capacitance and excellent rate performance. Through comprehensive analysis involving FESEM, BET, XRD, FTIR, XPS, and EIS, it was evident that the enhanced performance of NCH-2 is linked to its well-developed morphology, the presence of structural defects such as cobalt and oxygen vacancies, and superior charge transport dynamics. These findings highlight how subtle control of synthesis conditions, particularly pH, can lead to significant improvements in electrode material performance for energy storage devices.
Conflicts of interest
There are no conflicts to declare.
Data availability
Data are available upon request from the authors.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6ya00006a.
Acknowledgements
The author, Rajkamal Arya, gratefully acknowledges SRF fellowship support from the CSIR (Delhi) [22D/23J03561], and UPES SEED grant Project Code: UPES/R&D-SoAE/25062025/18. The authors express gratitude to CIC (UPES) for providing laboratories and characterisation facilities. IIT Ropar is thanked for its assistance with FESEM and XPS analysis, and RRCAT Indore is acknowledged for providing the Synchrotron XRD facility at Indus-2 (ADXRD beamline-12). Archna Sagdeo (RRCAT, Indore) is gratefully acknowledged for many useful discussions.
References
- P. Vaghela, V. Pandey, A. Sircar, K. Yadav, N. Bist and R. Kumari, MRS Energy Sustain., 2023, 10, 261–276 CrossRef.
- R. Kandpal and R. Singh, ECS Trans., 2022, 107, 8133 CrossRef CAS.
- K. Patil, P. Babar, D. M. Lee, V. Karade, E. Jo, S. Korade and J. H. Kim, Sustainable Energy Fuels, 2020, 4, 5254–5263 RSC.
- M. Sarfraz and I. Shakir, J. Energy Storage, 2017, 13, 103–122 CrossRef.
- S. K. Godlaveeti, A. El-marghany, R. R. Nagireddy, S. Gedi and R. Chintaparty, Ceram. Int., 2025, 51, 20620–20627 CrossRef CAS.
- R. Arya, T. Dagar and A. K. Sinha, Phys. Scr., 2025, 100, 35927 Search PubMed.
- B. Huang, W. Wang, T. Pu, J. Li, J. Zhu, C. Zhao, L. Xie and L. Chen, J. Colloid Interface Sci., 2018, 532, 630–640 CrossRef CAS PubMed.
- I. Irshad, A. Nazir, A. Tahir, A. G. Lone and B. Want, J. Energy Storage, 2025, 107, 114987 CrossRef CAS.
- A. Amin, M. Loewenich, L. Grebener, M. Hammad, S. Heckenbach, M.-A. Kräenbring, A. S. Odungat, A. H. Ladole, T. B. Nguyen, D. Schwabenland, H. K. Salim, H. Wiggers, D. Segets and F. Özcan, ACS Appl. Energy Mater., 2025, 8, 2050–2063 CrossRef CAS.
- Z. Sun, Z. Li, J. Peng, X. Yan, H. Shang, Y. Jin, Q. Zhao, C. Li, S. Lyu, C. Chen and J.-B. Baek, Energy Environ. Sci., 2025, 18, 5159–5189 Search PubMed.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef CAS PubMed.
- T. Zhao, H. Jiang and J. Ma, J. Power Sources, 2011, 196, 860–864 CrossRef CAS.
- M. Jana, P. Sivakumar, M. Kota, M. G. Jung and H. S. Park, J. Power Sources, 2019, 422, 9–17 CrossRef CAS.
- J. Yin, G. Zhou, X. Gao, J. Chen, L. Zhang, J. Xu, P. Zhao and F. Gao, Nanomaterials, 2019, 9, 1686 CrossRef CAS PubMed.
- M. S. Akhtar, T. Wejrzanowski, G. Komorowska, B. Adamczyk-Cieślak and E. Choinska, Electrochim. Acta, 2024, 508, 145284 CrossRef CAS.
- R. Arya, T. Dagar and A. K. Sinha, Ionics, 2025, 31, 2577–2591 CrossRef CAS.
- X. Yi, V. Celorrio, H. Zhang, N. Robertson and C. Kirk, J. Mater. Chem. A, 2023, 11, 22275–22287 Search PubMed.
- M. Bhatt, K. Gautam, A. Sagdeo and A. K. Sinha, ChemSusChem, 2025, 18, e202501348 CrossRef CAS PubMed.
- X. Wang, S. Wang, L. Ren, W. Wu, M. Zuo, W. Xing, B. Zhang, W. Fan, Z. He, Z. Yu, H. Zhang and W. Xiang, J. Alloys Compd., 2024, 977, 173458 Search PubMed.
- J. Hu, C. Wang, J. Cong, Y. Lan, G. Chen, C. Dong and H. Guan, J. Energy Storage, 2025, 139, 118826 CrossRef CAS.
- X.-Y. You, P.-Y. Lee, S.-C. Wang, C. Kongvarhodom, M. Saukani, S. Yougbaré, H.-M. Chen, K.-C. Ho, Y.-F. Wu and L.-Y. Lin, J. Energy Storage, 2024, 100, 113678 Search PubMed.
- T. Gao, X. Xiao, Z. Dong, X. Lu, L. Mao, J. Wang, Y. Liu, Q. Hu and J. Xu, Batteries, 2024, 10, 438 Search PubMed.
- M. Bhatt, B. Gupta and A. K. Sinha, Sci. Rep., 2025, 15, 2192 CrossRef CAS PubMed.
- A. K. Sinha, A. Sagdeo, P. Gupta, A. Upadhyay, A. Kumar, M. N. Singh, R. K. Gupta, S. R. Kane, A. Verma and S. K. Deb, J. Phys.: Conf. Ser., 2013, 425, 072017 Search PubMed.
- Y. Tang, Y. Liu, S. Yu, Y. Zhao, S. Mu and F. Gao, Electrochim. Acta, 2014, 123, 158–166 Search PubMed.
- J. Zhang, F. Liu, J. P. Cheng and X. B. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 17630–17640 Search PubMed.
- Y. Zhao, X. Zhang, J. He, L. Zhang, M. Xia and F. Gao, Electrochim. Acta, 2015, 174, 51–56 CrossRef CAS.
- T. Mathew, S. Srividhya, K. Venkatesh, L. Thomas and K. Nagaraju, Inorg. Chem. Commun., 2025, 174, 114074 Search PubMed.
- J. S. Lim and F. K. Yam, Phys. B, 2025, 699, 416798 CrossRef CAS.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- S. Pasieczna-Patkowska, M. Cichy and J. Flieger, Molecules, 2025, 30, 684 Search PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- S. Guo, Y. Wang, S. Xia, H. Li, S. Zuo and W. Xu, RSC Adv., 2025, 15, 11790–11798 RSC.
- M. Wang, Y. Hou, L. Yu and X. Hou, Nano Lett., 2020, 20, 6937–6946 Search PubMed.
- B. Shen, Y. He, W. Li, Z. Wang, L. Yu, Y. Jiang, X. Liu, J. Kang, H. Gao and N. Lin, Mater. Des., 2020, 191, 108645 Search PubMed.
- Q.-R. Pan, S.-J. Li, K. Tong, C. Xie, L. Peng, N. Li, D.-Y. Wang and H. Su, J. Mater. Sci., 2019, 54, 9063–9074 Search PubMed.
- O. A. Shlyakhtin, A. M. Skundin, S. J. Yoon and Y. J. Oh, Mater. Lett., 2009, 63, 109–112 CrossRef CAS.
- Z. Li, D. Yuan, S. Zhu, P. Fan, H. Ma, Q. Zhang, A. Wen and J. Zhu, J. Appl. Phys., 2019, 125, 175103 CrossRef.
- Y. Yuan, D. Chen, Y. Luo, T. Gao, C. Zhang, W. Zhang and Z. Yang, ACS Appl. Mater. Interfaces, 2024, 16, 60404–60414 Search PubMed.
- K. Bhunia, K. Serbara Bejigo and S.-J. Kim, Chem. Eng. J., 2024, 484, 149306 CrossRef CAS.
- Z. Cai, Y. Guo, C. Yang, Z. Li, S. Sun, M. Yue, X. Wang, M. Zhang, H. Wang, Y. Yao, D. Zheng, A. Farouk, F. A. Ibrahim, Y. Lv, X. Sun and B. Tang, Inorg. Chem. Front., 2025, 12, 154–160 Search PubMed.
- X. Zhang, Z. Yang, X. Cui, W. Liu, B. Zou and W. Liao, J. Colloid Interface Sci., 2022, 621, 1–11 Search PubMed.
- Y. Zhao, H. Li, P. Wang, S. Dong, Q. Wu, Q. Zhang and H. Zheng, J. Magn. Magn. Mater., 2023, 580, 170948 CrossRef CAS.
- X. Wei, Y. Chai, N. Liu, S. Qiao, Y. Fu and S. Chong, Int. J. Hydrogen Energy, 2022, 47, 9606–9615 CrossRef CAS.
- Y.-L. T. Ngo, L. Sui, W. Ahn, J. S. Chung and S. H. Hur, Nanoscale, 2017, 9, 19318–19327 RSC.
- S. Muduli, S. K. Pati, T. K. Pani and S. K. Martha, J. Energy Storage, 2023, 66, 107339 Search PubMed.
- A. Ramesh, N. S. Gudipati, S. R. K. Vanjari and Ch Subrahmanyam, Electrochim. Acta, 2023, 461, 142692 CrossRef CAS.
- X. Ren, L. Ren, Y. Hu, H. Sun, X. Han, C. Xu and H. Chen, Colloids Surf., A, 2024, 703, 135395 CrossRef CAS.
- Z. Li, C. Shi, J. Yu and L. Bai, Appl. Surf. Sci., 2024, 669, 160534 CrossRef CAS.
- S. Li, J. Qiu, C. Lai, M. Ling, H. Zhao and S. Zhang, Nano Energy, 2015, 12, 224–230 Search PubMed.
- F. Liu, C. Wu, Y. Dong, C. Zhu and C. Chen, J. Colloid Interface Sci., 2022, 628, 682–690 Search PubMed.
- J. Chen, J. Xu, S. Zhou, N. Zhao and C.-P. Wong, J. Mater. Chem. A, 2015, 3, 17385–17391 RSC.
- Y. X. Zhang, M. Huang, F. Li and Z. Q. Wen, Int. J. Electrochem. Sci., 2013, 8, 8645–8661 Search PubMed.
- H. Liu, J. Zhu, Z. Li, Z. Shi, J. Zhu and H. Mei, Chem. Eng. J., 2021, 403, 126325 Search PubMed.
- C. Xu, Y. Chen, Y. Ma, J. Huang, J. Zhao and H. Xu, Electrochim. Acta, 2021, 367, 137475 CrossRef CAS.
- G. Guo, Y. Mei, X. Chen, J. Liu and W. Liu, RSC Adv., 2023, 13, 25018–25028 RSC.
- A. Lasia, J. Phys. Chem. Lett., 2022, 13, 580–589 CrossRef CAS PubMed.
- C. Raminafshar, V. Dracopoulos, M. R. Mohammadi and P. Lianos, Electrochim. Acta, 2018, 276, 261–267 CrossRef CAS.
- R. Kumar, S. Tanwar, S. Kalia, Diksha, R. K. Singh and A. L. Sharma, Diamond Relat. Mater., 2026, 163, 113335 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Tanya Dagar
*a,
Tanya Dagar










