DOI:
10.1039/D5TC03810C
(Paper)
J. Mater. Chem. C, 2026,
14, 4646-4657
UVC-induced valence switching in BaFBr:Sm3+ nanoplates
Received
23rd October 2025
, Accepted 22nd January 2026
First published on 28th January 2026
Abstract
UVC-induced valence switching in nanocrystalline BaFBr:Sm3+ was studied in the context of photoexcitable storage phosphors. Nanocrystals were synthesized via hot-injection thermolysis of metal bromodifluoroacetates and consisted of square plates with average dimensions 34.4 nm (edge) × 4.9 nm (thickness); polydispersities in both dimensions were less than 10%. Nanoplates were found to undergo partial reduction upon exposure to UVC radiation of wavelength equal to 185 nm to yield BaFBr codoped with Sm3+ and Sm2+. Luminescence studies were conducted to probe a number of aspects of this photochemical reaction, including extent, kinetics, reversibility, and stability of photogenerated Sm2+ ions. Photoreduction extent depended on atmosphere (air vs. vacuum) and sample treatment (as-prepared vs. preheated), revealing the presence of photoactive centers at or near the surface of the nanoplates. Photoreduction kinetics was rationalized invoking a bimodal distribution of photoactive centers in which electron-trapping Sm3+ ions were distinguished on the basis of their distance to hole-trapping oxide defects. Valence switching was found to be reversible. Slow ionization of Sm2+ occurred in the dark via tunneling. Additionally, Sm2+ could be photoionized using light of appropriate wavelength via injection into the host's conduction band. Wavelength-dependent photoionization studies enabled construction of a host-referred binding energy diagram of photogenerated Sm2+ in BaFBr. Findings reported herein enlarge the library of photoexcitable storage nanophosphors based on alkaline-earth fluorohalides.
Introduction
Storage phosphors are photosensitive materials in which information may be written, read, and erased using light. Two types of phosphors ought to be distinguished: photostimulable and photoexcitable. Photostimulable phosphors are well-known and ubiquitous in X-ray radiography. In these phosphors, electron–hole pairs created upon interaction with X-rays are trapped in metastable centers thus creating a latent image.1 An intrinsic limitation of these materials as optical storage media is the destructive nature of the readout process, which entails emptying electron and hole traps. By contrast, photoexcitable phosphors are a class of storage phosphors in which exposure to X-ray or UVC radiation leads to stable luminescent centers.2–10 This allows for nondestructive readout by probing their photoluminescence response. This is a key advantage of photoexcitable phosphors that underpins their potential as optical storage media in dosimetry,2,5,6,8,11 multilevel data storage,3,9,12 and anticounterfeiting.13
Rare-earth-doped alkaline earth fluorohalides of formula MFX:RE (M = Ca, Sr, Ba; X = Cl, Br, I; RE = rare-earth) are well-established photostimulable phosphors. BaFBr:Eu2+, for example, has been the archetypical X-ray storage phosphor for more than four decades. More recent is the demonstration that this family of materials may serve as photoexcitable phosphors.2,14 Storage relies on reversible valence switching of a trivalent rare-earth activator doped into the fluorohalide host. Photoreduction of the rare-earth to its divalent state (RE3+ → RE2+) occurs upon exposure to X-ray or UVC radiation and constitutes a write operation. A read operation is performed by probing the photoluminescence of radiation-generated ions ([RE2+]* → RE2+), which may be excited using UVA/B or visible light. Finally, an erase operation entails using high-power illumination to bleach divalent ions via photoionization (RE2+ → RE3+). The functionality of alkaline-earth fluorohalides as photoexcitable storage phosphors relies on two distinct chemical units; these are electron-trapping trivalent rare-earth ions and hole-trapping oxide defects.3,4 Unlike rare-earth doping, oxygen doping is unintentional but unavoidable.15–18 Oxygen enters the anionic substructure as oxygen–halogen vacancy pairs, which result from substitution for fluorine or halogen and creation of a charge-compensating halide vacancy  .15,18–21 Negatively charged oxide defects are able to store holes,15,16,19 while positively charged halide vacancies may serve as electron traps (i.e., F color centers F(X−)). Although the involvement of color centers in the storage mechanism of photostimulable phosphors is well established,15,22–24 their role in the dynamics of electron trapping in photoexcitable phosphors remains an open question.4,5,8,25
.15,18–21 Negatively charged oxide defects are able to store holes,15,16,19 while positively charged halide vacancies may serve as electron traps (i.e., F color centers F(X−)). Although the involvement of color centers in the storage mechanism of photostimulable phosphors is well established,15,22–24 their role in the dynamics of electron trapping in photoexcitable phosphors remains an open question.4,5,8,25
To the best or our knowledge, the library of alkaline-earth fluorohalides that have been shown to serve as photoexcitable storage phosphors is limited to BaFCl doped with Sm3+. Riesen and coworkers have generated a body of research centered on the X-ray- and UVC-induced valence switching in that system,2,3,7,12,13,25–28 even though they also reported the feasibility of using Tm3+ as a photoreducible dopant.4 Likewise, studies from Zhang's group have focused on BaFCl:Sm3+, although in this case valence manipulation was accomplished through femtosecond-pulsed IR radiation.29–31 The focus on BaFCl as a host likely stems from the fact that this was the composition first studied by Riesen and Kaczmarek in 2007.2 The choice of trivalent samarium as an activator and electron trap is due to historical reasons too—BaFCl:Sm has been extensively studied as a spectral hole burning material32—but also to the distinct electronic structure of its divalent ion. Dieke diagrams for Sm3+ and Sm2+ are given in Fig. 1. The close proximity of Sm2+ excited states 4f6 (5DJ) and 4f55d1 directly affects photoluminescence and photoionization processes. Further, since the position of the 4f55d1 state depends on the local crystal field, the host's composition may be used as a lever to exercise synthetic control over those processes. Besides chemical composition, investigations from Riesen's and Zhang's groups shared the use of nanocrystalline BaFCl:Sm3+ prepared via coprecipitation as the photosensitive material. Strictly speaking, they used submicron-sized crystals featuring only one dimension in the nanoscale regime (<100 nm). Riesen's studies have indicated that transitioning from bulk to nanophosphors positively impacts a number of aspects of photosensitive response, including higher storage density (due to a higher concentration of defects) and reduced energy consumption (due to lower bleaching thresholds).2,3,12,26 However, the size dependence of this response remains to be established.
 |
| | Fig. 1 Dieke diagrams of Sm3+ and Sm2+. For the latter, the position of the lowest level of the 4f55d1 excited state corresponds to that derived in this work for BaFBr and overlaps with that of the 4f6 5D2 level (≈17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 cm−1, not shown). 800 cm−1, not shown). | |
Research opportunities that emerge from this brief review of the literature include expanding the library of photosensitive fluorohalide compositions and improving morphological control of nanocrystalline fluorohalides. As an example, BaFBr:Sm3+ is a remarkable absence in the library of photoexcitable phosphors, especially considering that its Eu2+-doped counterpart is the archetypical photostimulable phosphor. Likewise, colloidal routes to fluorohalide nanocrystals are absent from the synthetic toolbox, despite the fact that they could afford much better control over size and size distribution than coprecipitation. Expanding the compositional library and achieving morphological control would enlarge the knowledge base to derive composition–structure–photosensitivity while capitalizing functional advantages resulting from crystal size reduction. In this article, we leverage our group's expertise in the synthesis33 and structural analysis34 of nanocrystalline rare-earth-doped fluorohalides to address those two gaps in the field of photoexcitable storage phosphors. We report an investigation of the UVC-induced valence switching in monodisperse BaFBr:Sm3+ nanocrystals synthesized via hot-injection of metal difluorobromoacetates. Steady-state, time-resolved, and variable-temperature luminescence studies were conducted to characterize the nanophosphors’ photochemical response. The extent and kinetics of Sm3+ photoreduction and the stability of photogenerated Sm2+ were quantitatively analyzed with the aim of understanding the interplay between charge carrier traps, including the structural bases of this interplay. Energy levels of Sm2+ were mapped and their location within the bandgap of BaFBr estimated with an eye towards identifying optimum wavelengths to excite photoluminescence and induce photoionization.
Experimental
Synthesis of BaFBr:Sm3+ nanocrystals
Nanocrystal synthesis was conducted using standard Schlenk techniques under nitrogen atmosphere. BaCO3 (99.98%), Sm2O3 (99.9%), CF3COOH (99%), CF2BrCOOH (97%), oleic acid (90%), 1-octadecene (90%), and trioctylphosphine (97%) were used as reagents. All reagents were purchased from Sigma-Aldrich except CF2BrCOOH, which was purchased from Synquest Laboratories. Chemicals were used as received. CF3COOH, CF2BrCOOH, and trioctylphosphine were stored under N2. BaFBr:Sm3+ (4 mol%) nanocrystals were synthesized following a modified hot-injection route previously reported by our group.33 This approach entailed preparation of metal bromodifluoroacetate precursors followed by thermolysis in a mixture of high-boiling point organic solvents. Metal precursors were synthesized via solvent evaporation.35 BaCO3 (0.96 mmol) and Sm2O3 (0.02 mmol) were added to a 50 mL two-neck round-bottom flask containing 1 mL of CF3COOH and 5 mL of double-deionized H2O. The resulting mixture was heated at 65 °C for 12 h until a colorless and optically transparent solution was obtained. Then, 2 mmol of CF2BrCOOH were added to the solution and stirred until fully dissolved. Polycrystalline precursors were obtained by evaporating the solution at 65 °C for 48 h under a constant flow of nitrogen (200 mL min−1). Once evaporation was completed, 2 mL of oleic acid and 2 mL of octadecene were added to the flask containing the metal precursors. The flask was immersed in a sand bath and heated to 115 °C under vacuum for 45 min; magnetic stirring was used throughout. Then, the atmosphere was switched to nitrogen and the temperature of the sand bath increased to 140 °C. The solution was kept 5 min at that temperature and then injected into a 100 mL two-neck round-bottom flask containing 8 mL of trioctylphosphine at 300 °C under nitrogen atmosphere. A needle thermocouple was in direct contact with the reaction mixture. The temperature drop upon injecting all 4 mL of the precursor solution was ≈43–45 °C. The reaction proceeded for 20 min after injection. Then, the flask containing the reaction mixture was quenched to room temperature using a stream of air; a yellowish turbid solution was thus obtained. Nanocrystals were isolated by adding 80 mL of ethanol (200 proof), vortexing for 5 s, and then centrifuging at 8000 rpm for 10 min. The resulting precipitate was resuspended by adding a mixture of 7 mL of ethanol, 2 mL of methanol, and 5 mL of toluene and vortexing for 1 min. Centrifugation at 8000 rpm for 10 min and vacuum drying at room temperature concluded the work-up process. In a typical synthesis, ≈160–175 mg of polycrystalline BaFBr:Sm3+ were obtained (≈68–75% yield). The Sm/Ba molar ratio determined by ICP–OES (Galbraith Laboratories) was 0.0331, corresponding to a doping efficiency of 80%. Polycrystalline samples appeared off-white in color and were employed in chemical, structural, morphological, and luminescence studies.
Powder X-ray diffraction (PXRD)
PXRD patterns were collected using a Bruker D2 Phaser diffractometer operated at 30 kV and 10 mA. Cu Kα radiation (λ = 1.5418 Å) was employed. A nickel filter was used to remove Cu Kβ radiation. Diffractograms were collected in the 10–60° 2θ range using a step size of 0.012° and a step time of 1 s.
Rietveld analysis
Rietveld analysis36,37 of PXRD data was conducted using GSAS-II.38 The following parameters were refined: (1) scale factor and sample displacement; (2) background, which was modeled using a shifted Chebyschev polynomial function; (3) lattice constants; (4) atomic coordinates when allowed by space-group symmetry (P4/nmm); and (5) crystallite size and microstrain. Isotropic displacement parameters were fixed at 0.01, 0.03, and 0.02 Å2 for Ba, F, and Br, respectively. Difference curve and Rw residual were employed to assess the quality of the refined structural model.
Transmission electron imaging (TEM)
TEM images were obtained using a Thermo Scientific Talos F200X G2 S/TEM microscope operated at 200 kV. A small aliquot of the native solution containing BaFBr:Sm3+ nanocrystals was mixed with toluene and dropcast onto a 200 mesh Cu grid coated with a Lacey carbon film (Ted Pella Inc.). Size distribution histograms were obtained after analyzing 150 nanocrystals.
Spectrofluorometry
Spectrofluorometric analyses were conducted using a Fluorolog 3–222 fluorometer (Horiba Scientific). Steady-state spectra were collected using a 450 W xenon lamp and a variety of continuous-wave laser diodes (375, 405, 425, and 442 nm) as excitation sources. The 405 nm laser diode was operated in pulsed mode to collect time-resolved decays. A photomultiplier tube R928 served as the detector. UVC radiation from a 185 nm U-shaped lamp (49 uW cm−2 at 10 cm, Rexim, LLC) was used to induce photoreduction of Sm3+ to Sm2+. Photoreduction, photoluminescence, and photoionization experiments were conducted on polycrystalline BaFBr:Sm3+ loaded into a VPF–800 cryostat (Lake Shore Cryotronics), which allowed data collection at atmospheric and low pressures (<30 mTorr) and variable temperatures (80–800 K). The stage was equipped with a custom-made sample holder featuring a copper block with a 12 × 0.8 mm cavity (diameter × depth). The sample was held in place by a fluorescence-free UV-grade fused-silica window secured by a stainless-steel retainer and four spring-loaded screws. The UVC lamp was located outside the cryostat at ≈6 cm from the sample surface. Dynamic vacuum was monitored using a convection-enhanced Pirani gauge (Kurt J. Lesker, model KJL275807LL). Sample temperature was probed using a thermocouple directly connected to the copper holder and a cryogenic controller (Lake Shore Cryotronics, model 335 − 3060). Spectra and decays were collected with a bandpass of 2 nm.
Results and discussion
The phase purity and morphology of BaFBr:Sm3+ nanocrystals were probed using PXRD and TEM, respectively. A PXRD pattern and a representative TEM image are given in Fig. 2 along with nanocrystal size distribution histograms. Rietveld analysis confirmed the phase purity of polycrystalline BaFBr:Sm3+ as all diffraction maxima in the pattern were indexed to the fluorobromide phase (PDF No. 024–0090); no secondary crystalline phases were observed (Fig. 2a). Refined unit cell constants, atomic coordinates, and bond distances are given in the SI (see Table S1). Diffraction maxima exhibited anisotropic microstrain broadening that complicated adequate modeling of (101), (002), and (111) reflections (2θ ≈ 23.3°, 23.9°, and 30.7°, respectively). From a morphological standpoint, nanocrystals consisted of square plates with an average edge length of 34.4 nm and an average thickness of 4.9 nm (Fig. 2b and c; see Fig. S1 in SI for additional TEM images). Important in the context of this research is the observation that polydispersities below 10% were achieved in both dimensions. Such a narrow size distribution—i.e., monodisperse nanocrystals—is ideal to maximize the statistical significance of structure–photophysics and structure–photochemistry relationships derived from probing the nanocrystal ensemble. The size distribution reported herein for BaFBr:Sm3+ is significantly narrower than in our previous report of rare-earth-doped BaFBr nanocrystals synthesized via hot-injection, for which the polydispersity was 28%.33 We attribute this result to the combination of two synthetic modifications that delayed the beginning of Ostwald ripening (i.e., size defocusing); these were (i) injection of a larger volume of precursor solution (4 mL vs. 2 mL) at a much faster rate (2 s vs. 2 min), and (ii) heating the precursor solution to a higher temperature prior to injection (140 °C vs. 115 °C). Results from electron microscopy imaging confirmed the ability of hot-injection synthesis to afford significantly better morphological control than coprecipitation routes previously employed in the preparation of BaFCl:Sm3+. Although coprecipitation yielded plate-shaped fluorochloride crystals, their sizes were significantly larger than that of our monodisperse fluorobromide nanoplates (e.g., 300 × 200 × 70 nm3 and 600 × 400 × 90 nm3vs. 34 × 34 × 5 nm3);2,13,25 moreover, no information was provided regarding size distribution.
 |
| | Fig. 2 (a) Rietveld analysis of the PXRD pattern of BaFBr:Sm3+. Experimental (○) and calculated patterns ( ), difference curve ( ), difference curve ( , offset for clarity), and tick marks ( , offset for clarity), and tick marks ( ) corresponding to the calculated positions of the diffraction maxima are shown. (b) Representative TEM image showing nanoplates in two different orientations: base-lying and side-lying. (c) Size distribution histograms obtained after measuring 150 nanoplates. ) corresponding to the calculated positions of the diffraction maxima are shown. (b) Representative TEM image showing nanoplates in two different orientations: base-lying and side-lying. (c) Size distribution histograms obtained after measuring 150 nanoplates. | |
We then proceeded to screen the photoluminescence of as-prepared BaFBr:Sm3+ nanocrystals before and after exposure to UVC radiation. Results from those studies are summarized in Fig. 3. Excitation and emission spectra and time-resolved decays were collected at room temperature under ambient atmosphere. Bands arising from f–f radiative transitions of Sm3+ and from oxide defects were observed before UVC exposure (Fig. 3a). Sm3+-centered transitions appeared in the excitation spectrum (6H5/2 → 4F7/2, 402 nm) as well as in the emission spectrum (4G5/2 → 6HJ, J = 5/2 at 561 nm, 7/2 at 597 nm, 9/2 at 643 nm, and 11/2 at 702 nm). Emission bands sat on a broad background likely arising from oxide defects, whose emission peaked at 458 nm. Sm3+ emission was observed upon direct f–f excitation at 402 nm and upon charge-transfer excitation of oxide defects at 250 nm (O2− + Sm3+ (6H5/2) → O− + Sm2+ (7F0)). Oxide defects in BaFCl and BaFBr are well-known to absorb at 250 nm and below.3,15,18,20,39 Exposure of BaFBr:Sm3+ nanocrystals to UVC radiation (185 nm, 6.7 eV) for a few minutes led to partial photoreduction of Sm3+ to Sm2+, as demonstrated by the appearance of emission bands arising from f–f radiative transitions of the divalent ion (Fig. 3b, 5D0 → 7FJ, J = 0 at 688 nm, 1 at 703 nm, and 2 at 727 nm). The observation that valence switching occurred under sub-bandgap illumination (<8.3 eV)40,41 pointed to photoinduced electron transfer from oxide defects (hole traps) to Sm3+ (electron traps) as the photoreduction reaction, similar to what has been reported by Riesen and coworkers for BaFCl:Sm3+.3 This hypothesis was further supported by the fact that Sm3+ could be photoreduced upon exposure to 250 nm radiation (4.96 eV), which matches the absorption of oxide defects (see SI, Fig. S2). From a structural standpoint, photoinduced electron transfer relies on the close proximity of the donor and the acceptor. X-ray absorption studies conducted by our group demonstrated that trivalent rare-earth ions doped into alkaline-earth fluorohalides may indeed have oxide ions as nearest-neighbors;34 a similar scenario had been considered previously by Liu and coworkers on the basis of post-synthetic annealing studies of coprecipitated BaFCl:Sm3+.42 Sm2+ emission was observed upon 4f6 (7F0) → 4f55d1 excitation; the corresponding band extended from 285 to 550 nm and featured a maximum at 425 nm. The broadness of this band enabled simultaneous excitation of Sm2+ and Sm3+ at 402 nm or selective excitation of Sm2+ at 425 nm. The ability to excite both ions using a single wavelength close to 402 nm was important in the context of this research. As will be described below, we monitored the progress of photochemical reactions using two observables obtained upon laser excitation at 405 nm; these were emission intensities from Sm3+ (597 nm) and Sm2+ (688 nm) and their ratio. In fact, a 405 nm laser was used to collect time-resolved decays of Sm3+ at 597 nm (4G5/2 → 6H7/2) and of Sm2+ at 688 nm (5D0 → 7F0). Intensity-weighted average lifetimes (〈τ〉) of their corresponding excited states were computed according to eqn 1, where I(t) is the baseline-corrected luminescence intensity at time t, and ti and tf define the integration window.43
| | 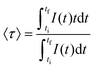 | (1) |
Integration was carried out from the first point of the decay until its return to the baseline level. Values of 2.83 and 1.63 ms were thus obtained for Sm
3+ and photogenerated Sm
2+, respectively (
Fig. 3c).
 |
| | Fig. 3 (a) and (b) Excitation and emission spectra of BaFBr:Sm3+ before and after exposure to UVC radiation under ambient atmosphere. Relevant transitions of Sm3+ and Sm2+ are labeled. (c) Time-resolved luminescence decays of Sm3+ and Sm2+ collected after UVC exposure. Intensity-weighted average lifetimes are given. | |
We decided to gain insight into the UVC-induced photochemistry of BaFBr:Sm3+ nanoplates. Specifically, we were interested in probing a number of aspects that are relevant from the standpoint of optical storage, including photoreduction extent, kinetics, and reversibility, as well as stability of photogenerated Sm2+ ions. To this end, a series of experiments was conducted, the first of which was to study the photoreduction of BaFBr:Sm3+ under ambient atmosphere (760 Torr, 22–25 °C, relative humidity = 22–26%) and under vacuum (5–30 mTorr, 22–25 °C). This experiment entailed following the photoreduction of Sm3+ under UVC illumination and the stability of the photogenerated Sm2+ in the dark as a function of time. Polycrystalline BaFBr:Sm3+ was loaded into the cryostat, exposed to 185 nm UVC radiation, and emission spectra were collected over time. Spectra were excited using a 405 nm laser diode (3.1 mW), thereby allowing simultaneous excitation of Sm2+ and Sm3+ and extraction of the ratio between the integrated intensities of their main emission bands at 688 and 597 nm (I688/I597), respectively; the UVC lamp was blocked during acquisition of spectra. Once saturation was achieved and the intensity ratio remained unchanged, the UVC lamp was turned off, and the sample left in the dark. Then, emission spectra were collected until the intensity ratio remained constant; this marked the completion of a UVC–dark cycle. BaFBr:Sm3+ solids were subjected to two consecutive UVC–dark cycles; a third exposure to UVC radiation was carried out to check repeatability. Intensity ratio vs. time curves were thus obtained and are given in Fig. 4; the time evolution of the corresponding integrated intensities is given in the SI (Fig. S3). Intensity ratio curves obtained under ambient atmosphere and under vacuum exhibited similar shape. The I688/I597 ratio—a qualitative proxy for the extent of photoreduction—increased under UVC illumination due to the photoreduction of Sm3+ and decreased in the dark due to detrapping of electrons from photogenerated Sm2+ (i.e., dark ionization). The time evolution of the ratio was driven by the intensity of Sm2+ emission, which evolved in the exact same way. However, there were two major differences between the photochemical response of BaFBr:Sm3+ under ambient atmosphere and under vacuum. The first difference involved the extent of photoreduction, which appeared to be larger under vacuum. A fivefold increase in the saturation intensity ratio was observed upon going from ambient (I688/I597 ≈ 0.50) to vacuum exposure (I688/I597 ≈ 2.50). The second difference involved the repeatability of the photochemical response, which was noticeably more consistent when conducting UVC–dark cycles under vacuum (i.e., the same saturation ratio was achieved three times). This difference stemmed from the evolution of Sm3+ emission, which was erratic under ambient atmosphere but repeatable under vacuum. Altogether, results from these studies provided two major lessons. The first lesson was that partial photoreduction of Sm3+ to Sm2+ was reversible and repeatable, more so under vacuum than under air. On this basis, we decided to conduct all subsequent experiments under dynamic vacuum (<30 mTorr). The second lesson was that at least a fraction of photosensitive Sm3+ ions was likely located at or near the surface of the nanocrystals. Thus, their ability to participate in photochemical reactions and photoluminesce was impacted by the presence of chemisorbed ligands. For example, one may envision a Sm3+ ion that was not photoactive under air due to the presence of coordinated water or hydroxyl groups, but became coordinatively unsaturated under vacuum and thus more prone to photoreduction. The participation of surface Sm3+ ions in photochemical reactions was further supported by the larger response obtained upon thermally treating BaFBr:Sm3+ nanocrystals under vacuum (vide infra). Infrared spectroscopy showed that, indeed, vacuum and thermal treatment of BaFBr:Sm3+ nanoplates induced partial dehydration of their surfaces as well as removal of residual organics (see SI, Fig. S4). For completeness, we add that X-ray absorption studies conducted by our group demonstrated that the local atomic environment of trivalent rare-earth ions doped into fluorohalide nanocrystals is highly disordered, compatible with a surface or subsurface location.34 Finally, the observation of electron detrapping in the dark raised the question of the kinetic stability of photogenerated Sm2+. Studies aimed at addressing this aspect are described in the following paragraph.
 |
| | Fig. 4 Luminescence intensity ratio curves obtained upon subjecting BaFBr:Sm3+ to UVC–dark cycles under ambient atmosphere and under vacuum. Ratios were computed by integrating emission bands at 597 (Sm3+) and 688 nm (Sm2+) between 582 and 614 nm and between 685 and 690 nm, respectively. Dashed and dotted lines are guides to the eye. UVC and dark exposure times are indicated. | |
Photoreduction under UVC illumination and ionization in the dark were further probed by monitoring the evolution of emission intensities over the course of a single UVC–dark cycle under vacuum. This study, however, featured two major differences with that described in the previous paragraph; these were (i) a larger number of emission spectra was acquired to get intensity vs. time curves suitable to extract kinetic information, and (ii) the 405 nm laser power was decreased to 0.5 mW to minimize optically induced detrapping (i.e., photoionization of Sm2+via excited-state tunneling to hole traps and/or through injection into the host's conduction band when allowed by trap depth). Completion of this experiment took ≈10 days and the corresponding results are given in Fig. 5; selected emission spectra are given in the SI (Fig. S5). The intensity ratio I688/I597 followed the expected trend, reaching a saturation value of ≈2.60 after 17 h of UVC exposure and decreasing to ≈1.40 after leaving the sample in the dark for 226 h (Fig. 5a). Similar to what was described earlier, this trend was driven by the evolution of Sm2+ emission at 688 nm. It should be noted that the intensity of this emission decreased ≈45% from its maximum value after ca. 9 days (Fig. 5b), thus limiting the use of our BaFBr:Sm3+ nanoplates as storage media. More importantly, the fact that extensive dark ionization was observed in monodisperse nanocrystals suggests that there may be a size limit to downscale photoexcitable storage phosphors without compromising functionality. With regards to Sm3+ emission at 597 nm, its evolution followed the expected trend during photoreduction, i.e., it decreased with UVC exposure time. By contrast, two stages were noticeable in the I597 curve extracted after ceasing UVC illumination. First, an increase in emission intensity was observed during the first 12 h in the dark, indicating that detrapping of electrons from Sm2+ generated luminescent Sm3+ ions. In the second stage, however, the intensity of Sm3+ emission remained practically constant even though dark ionization of Sm2+ occurred. Thus, detrapping took place without the generation of luminescent Sm3+ ions; a similar phenomenon was reported by Qin and coworkers in their study of photobleaching of BaF(Cl,Br):Sm2+.44 The time evolution of Sm2+ emission at 688 nm was quantitatively analyzed to extract rate constants for photoreduction and dark ionization. Previous kinetic studies of carrier trapping and detrapping in Sm-doped fluorides6,8 and fluorohalides3,25,27,44 had shown these processes to be dispersive. In the case of our BaFBr:Sm3+ nanoplates, we fitted the I688(t) curve using biexponential functions given in eqn (2) (photoreduction stage, t = 0–17 h) and eqn (3) (dark stage, t = 17–243 h).27 Here a is a scaling factor, f and (1 − f) are the fractions of the two distinct
| | Photoreduction: I688(t) = a{1 − f![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp[−k1(t − t0)] − (1 − f)exp[−k2(t − t0)]} exp[−k1(t − t0)] − (1 − f)exp[−k2(t − t0)]} | (2) |
| | | Dark: I688(t) = a{fexp[−k1(t − t0)] + (1 − f)exp[−k2(t − t0)]} | (3) |
Sm
2+ emitters,
k1 and
k2 their corresponding rate constants, and
t0 a time shift. Adequate fits were obtained for both stages (
Fig. 5b). Parameter values extracted for the photoreduction stage were
a = 1.019(3),
f = 0.794(7),
k1 = 0.229(4) h
−1, and
k2 = 2.0(1) h
−1. Values for the dark stage were
a = 0.969(7),
f = 0.221(8),
k1 = 0.09(1) h
−1, and
k2 = 0.00138(7) h
−1. These values revealed that Sm
2+ emission in both stages stemmed from a major slow component (≈80%) and a minor fast component (≈20%). This scenario was better visualized by plotting the individual contributions of each component (
Islow(
t) and
Ifast(
t)) to the total emission intensity (
I688(
t) =
Islow(
t) +
Ifast(
t)). These contributions were calculated using
eqn (4a) and (4b) (photoreduction stage) and
eqn (5a) and (5b) (dark stage).
| | | Photoreduction: Islow(t) = af[1 − exp(−k1t)] | (4a) |
| | | Ifast(t) = a(1 − f)[1 − exp(−k2t)] | (4b) |
| | | Dark: Islow(t) = a(1 − f)exp(−k2t) | (5a) |
| | | Ifast(t) = afexp(−k1t) | (5b) |
Inspection of
Islow(
t) and
Ifast(
t) curves thus derived showed that the photogeneration of Sm
2+ was dominated by the fast component for the first hour under UVC illumination (
Fig. 5c). At that point, the slow component became dominant and the fast component no longer contributed to the increase of the total emission intensity at 688 nm. Likewise, dark ionization was initially dominated by the fast component, which accounted for most of the ≈30% intensity drop observed within the first 46 h in the dark. At the end of that period, emission from the fast component was negligible and the decrease in the intensity at 688 nm was entirely due to the decay of the slow component. It is worth noting that fast detrapping from Sm
2+ coincided in time with the partial recovery of the Sm
3+ emission at 597 nm, whereas slow detrapping did not generate luminescent Sm
3+ ions. For completeness, we also note that the time evolution of the emission decay from the slow component was quasilinear within the time window considered (17–243 h). In fact, a linear fit of this curve yielded a fit residual equal to 0.998 and a decay rate equal to 0.089% h
−1 (0.999 and 0.086% h
−1, respectively, were obtained when the fit was conducted between 63 and 243 h). The biexponential kinetics of electron trapping (photoreduction) and detrapping (dark ionization) and the fact that the fractions of each component were the same in both processes pointed to a bimodal distribution of photoactive Sm
3+ centers within the nanocrystals.
 |
| | Fig. 5 Kinetics of UVC photoreduction and dark ionization of BaFBr:Sm3+ under vacuum. Time evolution of the luminescence intensity ratio (a) and of the corresponding integrated intensities I597 and I688 (b). A magnified view of the I597(t) curve is shown in the inset of (b). Fits of equations 2 and 3 to the I688(t) curve are shown as solid red and blue lines, respectively. Fit residuals are given. (c) I688(t) curve calculated according to eqn (4a) and (4b) (UVC stage) and eqn (5a) and (5b) (dark stage). Contributions from slow and fast components to the total intensity are plotted. Dashed and dotted lines are guides to the eye. | |
From a structural standpoint, two types of Sm3+ ions could be distinguished on the basis of their proximity to oxide defects. The first type of Sm3+ centers (type I) consisted of those having oxide defects in close proximity so that exposure to UVC radiation favored direct O2− → Sm3+ electron transfer and caused fast photoreduction of Sm3+. Dark ionization of the photogenerated Sm2+ occurred via direct electron backtransfer or short-distance tunneling and quickly restored the luminescence of the parent Sm3+. The local structure of type I Sm3+ was rather static since the same photochemical response was observed upon UVC–dark cycling. The second type of Sm3+ centers (type II) involved those having oxide defects at a longer distance, such that photoreduction via direct electron transfer was not feasible. It is worth recalling that oxygen typically enters the fluorohalide lattice as oxide–halide ion vacancy pairs and that these defect pairs may act as hole–electron traps under X-ray or UVC radiation.15,18 We propose that UVC exposure led to photoexcitation of these pairs via formation of O− and a neutral color center (F(X−)), followed by slow diffusion of the latter through the fluorohalide lattice to generate Sm2+via reaction with a type II Sm3+ . This reaction bleached Sm3+ centers that were initially luminescent. Photodissociation of oxide–halide ion vacancy pairs and diffusion of the resulting color centers is a key piece underlying the functionality of photostimulable BaFBr:Eu2+ phosphors.15,23,24 Diffusion of carrier traps was also brought up in studies of BaFCl:Sm3+,12 BaFCl:Tm3+,4 BaLiF3:Sm3+,6 and CaF2:Sm3+.8 It must be invoked here to rationalize the observation that the initial emission intensity from Sm3+ was not recovered after dark ionization following the first exposure to UVC radiation. In other words, the first exposure to UVC radiation caused an irreversible structural change that bleached Sm3+ centers that were initially luminescent. Thus, unlike the case of type I Sm3+, a dynamic structural component appeared to be involved in the initial photoreduction of type II Sm3+. Radiation-induced structural distortions were also observed in NaMgF3:Sm.45 Luminescent Sm2+ centers generated via diffusion of color centers underwent slow dark ionization to yield non-luminescent Sm3+ and regenerate distant O2− hole traps via long-distance tunneling (Sm2+ + O− → Sm3+ + O2−). For completeness we note that the bimodal distribution proposed here bears resemblance to that described by Eachus et al. for photostimulable centers in BaFBr:Eu2+,21 as both rely on a distribution of distances between carrier traps to rationalize experimental results.
. This reaction bleached Sm3+ centers that were initially luminescent. Photodissociation of oxide–halide ion vacancy pairs and diffusion of the resulting color centers is a key piece underlying the functionality of photostimulable BaFBr:Eu2+ phosphors.15,23,24 Diffusion of carrier traps was also brought up in studies of BaFCl:Sm3+,12 BaFCl:Tm3+,4 BaLiF3:Sm3+,6 and CaF2:Sm3+.8 It must be invoked here to rationalize the observation that the initial emission intensity from Sm3+ was not recovered after dark ionization following the first exposure to UVC radiation. In other words, the first exposure to UVC radiation caused an irreversible structural change that bleached Sm3+ centers that were initially luminescent. Thus, unlike the case of type I Sm3+, a dynamic structural component appeared to be involved in the initial photoreduction of type II Sm3+. Radiation-induced structural distortions were also observed in NaMgF3:Sm.45 Luminescent Sm2+ centers generated via diffusion of color centers underwent slow dark ionization to yield non-luminescent Sm3+ and regenerate distant O2− hole traps via long-distance tunneling (Sm2+ + O− → Sm3+ + O2−). For completeness we note that the bimodal distribution proposed here bears resemblance to that described by Eachus et al. for photostimulable centers in BaFBr:Eu2+,21 as both rely on a distribution of distances between carrier traps to rationalize experimental results.
Kinetic studies of electron trapping and detrapping were followed by a series of experiments aimed at mapping the energy levels of photogenerated Sm2+ centers. Specifically, we sought to establish the location of the 4f55d1 excited state relative to the 4f6 7F0 ground state and to the bottom of the host's conduction band. The 4f55d1 excited state is directly involved in the photophysical and photochemical response of Sm2+ because it (i) acts as a feeding state for 4f6 5DJ levels and impacts 5DJ → 7F0 radiative processes through 4f–5d electronic mixing,46–51 (ii) participates in the nonradiative depopulation of 5DJ levels through thermally activated 5DJ → 4f55d1 crossover,46,48–50 and (iii) impacts the stability of photogenerated Sm2+ by serving as an intermediate state during photoionization (via ground-state absorption followed by excited-state tunneling to hole traps or through excited-state absorption to the conduction band)3,52 and photothermal ionization (ground state absorption followed by thermal injection into the conduction band).53 Our strategy to establish the position of the 4f55d1 excited state relative to the 7F0 ground state was that described by Pal et al.50 and Shen and Bray.49 This approach entailed probing and modeling the temperature dependence of the lifetime corresponding to the 5D1 → 7F0 radiative decay at 629 nm (15898 cm−1). To this end, steady-state emission spectra and time-resolved decays excited at 405 nm were collected between 80 and 300 K under vacuum (<30 mTorr). For these experiments, we used BaFBr:Sm3+ nanocrystals that had been previously saturated under UVC illumination and subsequently left in the dark for several days (until I688/I597 ≈ 1.6). The corresponding results are given in Fig. 6. Emission from the 5D1 → 7F0 transition could be observed in the entire temperature range (Fig. 6a). The corresponding peak at 629 nm sat on a decreasing background. At room temperature, its intensity was very weak and it appeared as a shoulder of the Sm3+ 4G5/2 → 6H9/2 emission band centered at 642 nm. Time-resolved decays collected in 20 K increments were fit using a triexponential function given in eqn (6). Here I(t) is the luminescence intensity at time t, τi is the lifetime of i-th component, and Ai and A are constants. Fractional contributions
| |  | (6) |
| |  | (7) |
| |  | (8) |
Fi were computed according to
eqn (7) and the intensity-weighted average lifetime 〈
τ〉 estimated using
eqn (8). In this case, a multiexponential approach was preferred over integration because of the need to go beyond the average picture in order to extract the temperature dependence of the
5D
1 excited-state lifetime. Experimental decays and fits are given in the SI (Fig. S6). Decays were dominated by a major component with lifetimes in the ≈6–9 ms range (
τ1, see
Fig. 6b). These values, however, were significantly larger than those expected for the
5D
1 excited state of Sm
2+ doped into fluorohalide hosts, which should be in the submilisecond range.
46,49,50 In the case of the second component (
τ2), lifetime values ranged between ≈0.7 and 1.7 ms and did not show the temperature dependence expected for the
5D
1 excited state, whose lifetime is known to decrease with temperature until reaching a few hundreds or tens of microseconds near room temperature.
46,49,50 On the other hand, lifetimes extracted for the third component (
τ3) were compatible with those expected for the
5D
1 excited state, both in terms of magnitude and temperature dependence; thus,
τ3 values are denoted
τ(
5D
1) hereafter.
τ(
5D
1) ranged from 140 µs at 80 K to 23 µs at 300 K; lifetime values were constant up to 180 K, where the onset of thermal quenching was observed (
Fig. 6c). Although Pal
et al. observed the onset of thermal quenching at a similar temperature (
ca. 200 K), the shape of their
τ(
5D
1)
vs. temperature curve differed from ours. Three temperature regimes were clearly noticeable in their case: constant lifetimes below 100 K (≈800 µs) were followed by a moderate decrease up 200 K (≈700 µs) and exhibited a drastic decrease upon going to 300 K (≈40 µs).
50 A qualitatively similar three-temperature regime was reported by Shen and Bray for SrFCl:Sm
2+.
49 Quantitative insight into the temperature dependence of the
5D
1 level was gained by fitting
eqn (9) to the
| |  | (9) |
experimental
τ(
5D
1)(
T) curve. This model accounts for depopulation of the excited state
via one radiative channel (
5D
1 →
7F
0 emission) and two nonradiative channels (
5D
1 →
5D
0 multiphonon relaxation and
5D
1 → 4f
55d
1 thermally-activated crossover).
46,49,50,54 Here,
KR is the radiative constant,
K10 is the spontaneous multiphonon emission rate, Δ
E10 is the energy gap between the
5D
1 and
5D
0 levels, and
p is the number of phonons needed to bridge that gap. In the crossover term,
K1fd is a frequency factor and Δ
E1fd is the energy gap between the
5D
1 level and the lowest level of the 4f
55d
1 excited state. The best fit was obtained by setting
KR = 6897 s
−1 (based on a radiative lifetime equal to 145 µs), Δ
E10 = 1363 cm
−1, and
p = 5.7 (based on an effective phonon frequency equal to 240 cm
−1).
55 Doing so, yielded
K10 = 0(50) s
−1,
K1fd = 1.97(1.80) × 10
8 s
−1, and Δ
E1fd = 1768(163) cm
−1 as refined parameters and
r2 = 0.994 as the fit residual. The zero value obtained for the multiphonon relaxation constant
K10 reflected the negligible contribution of this nonradiative process to the depopulation of the
5D
1 level; a similar result was reported by He
et al. in their study of BaCl
2:Sm
2+.
54 In the case of Pal
et al.'s study of BaFBr:Sm
2+, inclusion of the multiphonon relaxation term was needed to adequately model the three temperature regimes observed in the
τ(
5D
1)(
T) curve.
50 The value of 1768 cm
−1 obtained for the
5D
1–4f
55d
1 gap implied that the lowest level of 4f
55d
1 excited state of Sm
2+ was located 3131 cm
−1 (0.39 eV) above the
5D
0 level and 17
666 cm
−1 (2.19 eV) above the
7F
0 ground state. This estimate was in excellent agreement with that reported by Cui and coworkers based on variable-temperature steady-state spectra of BaFBr:Sm
2+ (17
860 cm
−1, 2.21 eV)
56 but lower than that reported by Pal and coworkers (18
469 cm
−1, 2.29 eV).
50
 |
| | Fig. 6 (a) Emission spectra of BaFBr:Sm2+/3+ at 80 and 300 K. Emission from the 5D1 → 7F0 decay of Sm2+ at 629 nm is depicted with the * symbol. (b) Results from triexponential fits of variable-temperature time-resolved luminescence decays at 629 nm. Lifetimes and fractional contributions (%) are given for each component along with the intensity-weighted average lifetime. τ3 corresponds to the lifetime of the 5D1 level of Sm2+ (τ(5D1)). Dotted lines are guides to the eye. (c) Temperature dependence of τ(5D1). Fit of eqn (9) to experimental lifetimes is depicted as a solid red line. Fit residual is given. | |
The energy of the 4f55d1 excited state of Sm2+ relative to the 7F0 ground state was subsequently used to estimate the gap between the 4f55d1 state and the bottom of the host's conduction band (CB). This was accomplished by irradiating BaFBr:Sm2+/3+ nanocrystals with laser light of decreasing energy until photoionization of Sm2+ was no longer observed due to the inability to bridge the 7F0–CB gap. In these experiments, BaFBr:Sm3+ was first exposed to UVC radiation to generate Sm2+ (I688/I597 ≈ 3.20), and then continuously to laser light of fixed wavelength for 2 h. 375, 405, and 442 nm were used as wavelengths (λexposure), corresponding to energies equal to 3.31, 3.06, and 2.81 eV, respectively; in all three cases, the laser power was fixed at 0.5 mW. Emission spectra were collected before and after laser exposure using 405 nm excitation (0.5 mW). Photoionization experiments were conducted under vacuum using BaFBr:Sm3+ nanocrystals that had been thermally treated at 450 K for 6 h in the cryostat. These nanocrystals exhibited a photochemical response that was qualitatively similar to their as-prepared counterparts (see SI, Fig. S7 and S8). However, from a quantitative standpoint, two differences should be pointed out. First, the extent of photoreduction appeared to be larger in thermally treated samples judging from the saturation intensity ratio (I688/I597 ≈ 3.70 vs. 2.50 in as-prepared nanocrystals). Additionally, dark ionization was slower judging from the percent change in the Sm2+ emission intensity (ΔI688 ≈ –4% in the first 2 h after ceasing UVC exposure vs. –9% in as-prepared nanocrystals). The increase in reaction extent upon going from as-prepared samples under air and vacuum to thermally treated samples confirmed that at least a fraction of the photoactive Sm3+ centers was located at or near the nanocrystals’ surface. Slower dark ionization enabled probing photoionization of a larger population of Sm2+ centers. For completeness, we note that PXRD and thermal analyses demonstrated that treatment at 450 K did not impact the structural integrity of the nanocrystals (see SI, Fig. S9). Results from photoionization experiments are summarized in Fig. 7. Emission spectra shown therein are baseline-subtracted; raw spectra are given in the SI (Fig. S10). Exposure to 375 and 405 nm light caused a decrease of 47 and 30%, respectively, in the integrated intensity of the 688 nm band of Sm2+. Thus, ionization of Sm2+via excitation into the conduction band occurred in these cases. By contrast, the decrease observed upon exposure to 442 nm light was comparable to that observed in the dark (ΔI688 ≈ –5%), indicating that no light-induced detrapping took place. On this basis, the upper and lower limits for the 4f55d1–CB gap were estimated using the energies corresponding to 405 and 442 nm photons as trap depths and that of the 7F0–4f55d1 gap (2.19 eV); values of 0.87 (3.06–2.19) and 0.62 (2.81–2.19) eV were thus obtained.
 |
| | Fig. 7 Emission spectra of BaFBr:Sm2+/3+ before and after exposure to laser light of wavelength λexposure. Spectra were collected using 405 nm excitation. The percent change of Sm2+ emission intensity at 688 nm (ΔI688) is given. | |
Energy differences were used to construct a host-referred binding energy (HRBE) diagram of photogenerated Sm2+ in BaFBr:Sm3+ nanoplates; it is given in Fig. 8. The diagram was built using a value of 8.3 eV for the bandgap of BaFBr40,41 and 0.87 eV for the 4f55d1–CB gap. The 7F0 ground state of Sm2+ was placed 2.19 eV below the 4f55d1 excited state. We tentatively placed oxide defect levels 4.96 eV below Sm2+ 7F0 level based on the observation of an O2− → Sm3+ charge-transfer band at 250 nm; the energy of this excitation provided a means of estimating the ground state of photogenerated Sm2+ ions. Color centers were not included in the HRBE diagram but literature reports had placed them ≈2–2.5 eV below the bottom of BaFBr's conduction band.22,57,58 The diagram is an average depiction of the position of energy levels within the host's bandgap. Nonetheless, it is informative from the standpoint of exploring the functionality of BaFBr:Sm3+ nanoplates as optical storage media. Specifically, it provides insight into how to approach reading and erasing Sm2+ centers generated upon UVC exposure. With regards to reading, one ideally wants to photoexcite Sm2+ without causing bleaching. From this perspective, results presented herein show that using light of wavelength longer than 405 nm is desirable if one is interested in probing Sm2+ emission intensities only. If excitation at 405 nm is used because the Sm2+/Sm3+ luminescence intensity ratio is of interest, then very low powers (≪0.5 mW) should be employed to minimize optically induced detrapping. In the case of erasing, single-photon photoionization may be achieved using light of wavelength shorter than or equal to 405 nm. Increasing optical power increases the extent of photobleaching. As an example, a 72% decrease in the Sm2+ emission intensity was achieved by exposing BaFBr:Sm2+/3+ nanocrystals to 405 nm light of increasing power (up to 50 mW, see SI, Fig. S11). Multiphoton photoionization should also be possible using light of wavelength longer than 405 nm and 4f6 (5DJ) and 4f55d1 excited states as intermediates.3,29 Finally, based on the HRBE diagram, one may envision photothermal ionization as an alternative for bleaching Sm2+ centers.53 In principle, this could be accomplished by coupling ≈560 nm (2.2 eV) or shorter wavelength excitation to temperature. Then, optical population of the 4f55d1 excited state would be followed by thermal depopulation via injection into the conduction band. This approach, however, could be limited by the highest temperature achievable without compromising the chemical and structural integrity of the nanoplates.
 |
| | Fig. 8 Host-referred binding energy diagram of photogenerated Sm2+ in BaFBr:Sm3+. The diagram was constructed using a value of 8.3 eV for the bandgap of BaFBr and of 0.87 eV for the 4f55d1–CB gap (see text for details). | |
Conclusions
In summary, we showed that monodisperse BaFBr:Sm3+ nanoplates undergo reversible valence switching upon exposure to UVC radiation. Sm3+ and oxide defects served as electron and hole traps, respectively. The kinetics of photoreduction and dark ionization processes was rationalized invoking a bimodal distribution of photoactive Sm3+ centers, in which these were distinguished on the basis of their distance to oxide defects. One of these types of Sm3+ ions was permanently bleached after the first exposure to UVC radiation. We proposed that this was due to an irreversible structural change connected to the diffusion of color centers, which acted as transient electron traps. Results from photoionization studies served to estimate the depth of electron traps, which were found to be located between ≈2.8 and 3.1 eV below the bottom of the BaFBr host's conduction band.
This article contributes to close two gaps in the field of photoexcitable storage nanophosphors because it unveils the photochemical response of a previously unexplored fluorohalide composition and does so using nanocrystals with uniform morphology. Avenues for future work include (i) gaining further synthetic control over nanocrystal average size to probe the size-dependence of photoreduction and dark ionization extents, which impact storage density and stability; and (ii) probing the structural evolution of electron and hole traps during photoreduction and dark ionization to conclusively establish the spatial distribution of these centers and their dynamics.
Conflicts of interest
There are no conflicts to declare.
Data availability
All the data used are provided in the article and the supplementary information (SI). Supplementary information: (1) additional results from X-ray diffraction and electron microscopy analyses, (2) results from thermal analyses, and (3) additional results from luminescence studies. See DOI: https://doi.org/10.1039/d5tc03810c.
Acknowledgements
The authors would like to acknowledge the financial support of the National Science Foundation (DMR−2508002) and of the Department of Chemistry at Wayne State University. They also thank the Lumigen Instrument Center at Wayne State University for the use of the powder diffractometer (National Science Foundation MRI–1427926) and of the transmission electron microscope (National Science Foundation MRI–2018587).
Notes and references
- K. Takahashi, J. Miyahara and Y. Shibahara, Photostimulated Luminescence (PSL) and Color-Centers in BaFCl-Eu2+, BaFBr-Eu2+, BaFI-Eu2+ Phosphors, J. Electrochem. Soc., 1985, 132, 1492–1494 CrossRef CAS
 .
.
- H. Riesen and W. A. Kaczmarek, Efficient X-ray Generation of Sm2+ in Nanocrystalline BaFCl/Sm3+: A Photoluminescent X-ray Storage Phosphor, Inorg. Chem., 2007, 46, 7235–7237 CrossRef CAS PubMed .
- H. Riesen, K. Badek, T. M. Monro and N. Riesen, Highly Efficient Valence State Switching of Samarium in BaFCl:Sm Nanocrystals in the Deep UV for Multilevel Optical Data Storage, Opt. Mater. Express, 2016, 6, 3097–3108 CrossRef CAS .
- J. Zhang and H. Riesen, Controlled Generation of Tm2+ Ions in Nanocrystalline BaFCl:Tm3+ by X-ray Irradiation, J. Phys. Chem. A, 2017, 121, 803–809 CrossRef CAS PubMed .
- J. J. Schuyt and G. V. M. Williams, Radiation-Induced Changes in the Optical Properties of NaMgF3(Sm): Observation of Resettable Sm Radio-Photoluminescence, Mater. Res. Bull., 2018, 106, 455–458 CrossRef CAS .
- N. Chowdhury, N. Riesen and H. Riesen, Efficient Generation of Stable Sm2+ in Nanocrystalline BaLiF3:Sm3+ by UV- and X-Irradiation, J. Phys. Chem. C, 2019, 123, 25477–25481 CrossRef CAS .
- J. Zhang and H. Riesen, Photostimulated and Persistent Luminescence of Samarium Ions in BaFCl, J. Lumin., 2019, 207, 188–194 CrossRef CAS .
- Z. S. Rozaila, N. Riesen and H. Riesen, Luminescence and Photoionization of X-ray Generated Sm2+ in Coprecipitated CaF2 Nanocrystals, Dalton Trans., 2021, 50, 16205–16213 Search PubMed .
- N. Wahib, N. Riesen and H. Riesen, Reversible Mn Valence State Switching in Submicron α-Al2O3:Mn by Soft X-rays and Blue Light – A Potential Pathway Towards Multilevel Optical Data Storage, Phys. Chem. Chem. Phys., 2022, 24, 6155–6162 RSC .
- J. J. Schuyt, G. V. M. Williams and S. V. Chong, Optically Reversible Tm3+ → Tm2+ Radiophotoluminescence in NaMgF3:Tm, Opt. Mater., 2022, 133, 112926 Search PubMed .
- G. Okada, Y. Koguchi, T. Yanagida, S. Kasap and H. Nanto, Recent Advances in Radiophotoluminescence Materials for Luminescence Dosimetry, Jpn. J. Appl. Phys., 2023, 62, 010609 CrossRef CAS .
- N. Riesen, X. Pan, K. Badek, Y. Ruan, T. M. Monro, J. Zhao, H. Ebendorff-Heidepriem and H. Riesen, Towards Rewritable Multilevel Optical Data Storage in Single Nanocrystals, Opt. Express, 2018, 26, 12266–12276 Search PubMed .
- N. Riesen, C. Priest, D. G. Lancaster, K. Badek and H. Riesen, Ultra-High-Resolution Greyscale Fluorescence Images via UV-Exposure of Thin Flexible Phosphor Films, Nanoscale, 2023, 15, 4863–4869 RSC .
- M. Matsarski, D. Lovy, H. Bill and K. M. Mohnhaupt, Light-Induced Valence-State Switching in BaFCl:La and SrFCl:La, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 205113 Search PubMed .
- R. S. Eachus, W. G. McDugle, R. H. D. Nuttall, M. T. Olm, F. K. Koschnick, T. Hangleiter and J. M. Spaeth, Radiation-Produced Electron and Hole–Centers in Oxygen-Containing BaFBr: 1. EPR and ODEPR Studies, J. Phys.: Condens. Matter, 1991, 3, 9327–9338 CrossRef CAS .
- R. S. Eachus, W. G. McDugle, R. H. D. Nuttall, M. T. Olm, F. K. Koschnick, T. Hangleiter and J. M. Spaeth, Radiation-Produced Electron and Hole–Centers in Oxygen-Containing BaFBr: 2. An ENDOR Study of O−(F), J. Phys.: Condens. Matter, 1991, 3, 9339–9349 CrossRef CAS .
- T. J. Bastow, S. N. Stuart, W. G. McDugle, R. S. Eachus and J. M. Spaeth, Oxygen Impurities in X-ray Storage Phosphors BaFBr and BaFCl Investigated by 17O NMR, J. Phys.: Condens. Matter, 1994, 6, 8633–8644 CrossRef CAS .
- F. K. Koschnick, T. Hangleiter, K. S. Song and J. Spaeth, Optically Detected Magnetic Resonance Study of an Oxygen–Vacancy Complex in BaFBr, J. Phys.: Condens. Matter, 1995, 7, 6925–6937 CrossRef CAS .
- R. C. Baetzold, Calculation of Properties of Charged Defects in BaFBr and SrFCl, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 7994–8002 CrossRef CAS PubMed .
- E. Radzhabov and V. Otroshok, Optical Spectra of Oxygen Defects in BaFCl and BaFBr Crystals, J. Phys. Chem. Solids, 1995, 56, 1–7 CrossRef CAS .
- R. S. Eachus, R. H. D. Nuttall, M. T. Olm, W. G. McDugle, F. K. Koschnick, T. Hangleiter and J. M. Spaeth, Oxygen Defects in BaFBr and BaFCl, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 3941–3950 CrossRef CAS PubMed .
- H. von Seggern, T. Voigt, W. Knüpfer and G. Lange, Physical Model of Photostimulated Luminescence of X-ray Irradiated BaFBr:Eu2+, J. Appl. Phys., 1988, 64, 1405–1412 CrossRef CAS .
- F. K. Koschnick, J. M. Spaeth, R. S. Eachus, W. G. McDugle and R. H. D. Nuttall, Experimental Evidence for the Aggregation of Photostimulable Centers in BaFBr:Eu2+ Single Crystals by Cross Relaxation Spectroscopy, Phys. Rev. Lett., 1991, 67, 3571–3574 CrossRef CAS PubMed .
- F. K. Koschnick, J. M. Spaeth and R. S. Eachus, The Influence of Oxide Impurity on the Generation by X-Irradiation of F Centres in BaFBr, J. Phys.: Condens. Matter, 1992, 4, 3015–3029 Search PubMed .
- Z. Q. Liu, M. Stevens-Kalceff and H. Riesen, Photoluminescence and Cathodoluminescence Properties of Nanocrystalline BaFCl:Sm3+ X-ray Storage Phosphor, J. Phys. Chem. C, 2012, 116, 8322–8331 CrossRef CAS .
- M. A. Stevens-Kalceff, Z. Liu and H. Riesen, Cathodoluminescence Microanalysis of Irradiated Microcrystalline and Nanocrystalline Samarium Doped BaFCl, Microsc. Microanal., 2012, 18, 1229–1238 CrossRef CAS PubMed .
- N. Riesen, A. François, K. Badek, T. M. Monro and H. Riesen, Photoreduction of Sm3+ in Nanocrystalline BaFCl, J. Phys. Chem. A, 2015, 119, 6252–6256 CrossRef CAS PubMed .
-
N. Riesen, K. Badek, L. T. Kasim, Y. Ruan, T. M. Monro and H. Riesen, Multilevel Optical Data Storage using Samarium-Doped Matlockite Nanocrystals, 2017 IEEE Photonics Conference (IPC), 2017, pp. 599–600 Search PubMed.
- Z. Pan, L. Z. Deng, L. Cheng, Y. H. Yao, D. L. Qi, W. J. Cheng, Z. R. Sun and S. Zhang, Multiphoton Induced Photoreduction in BaFCl:Sm3+ Nanocrystals Under Femtosecond Laser Field, J. Lumin., 2023, 263, 120052 Search PubMed .
- Z. Pan, B. Zheng, L. Deng, Y. Yao, D. Qi, Y. Shen, Z. Sun and S. Zhang, Spectral Phase-Based Valence State Conversion Manipulation in BaFCl:Sm3+ Nanocrystals for Multilevel Information Recording and Display, Opt. Express, 2024, 32, 43643–43653 Search PubMed .
- B. Zheng, L. Z. Deng, J. Li, Y. H. Yao, D. L. Qi, Y. C. Shen, Z. R. Sun and S. A. Zhang, Single Femtosecond Laser Pulse-Induced Valence State Conversion in BaFCl:Sm3+ Nanocrystals for Low-Threshold Optical Storage, Nanophotonics, 2024, 13, 3709–3715 CrossRef CAS PubMed .
- A. Winnacker, R. M. Shelby and R. M. Macfarlane, Photon-Gated Hole Burning: A New Mechanism Using Two-Step Photoionization, Opt. Lett., 1985, 10, 350–352 CrossRef CAS PubMed .
- B. D. Dhanapala, H. N. Munasinghe, K. T. Dissanayake, L. Suescun and F. A. Rabuffetti, Expanding the Synthetic Toolbox to Access Pristine and Rare-Earth-Doped BaFBr Nanocrystals, Dalton Trans., 2021, 50, 16092–16098 RSC .
- D. K. Amarasinghe, K. T. Dissanayake, B. D. Dhanapala and F. A. Rabuffetti, Local Atomic Environment of Yb3+ in Alkaline-Earth Fluorohalide Nanocrystals, CrystEngComm, 2022, 24, 5317–5323 RSC .
- K. T. Dissanayake, L. M. Mendoza, P. D. Martin, L. Suescun and F. A. Rabuffetti, Open-Framework
Structures of Anhydrous Sr(CF3COO)2 and Ba(CF3COO)2, Inorg. Chem., 2016, 55, 170–176 CrossRef CAS PubMed .
- H. M. Rietveld, Line Profiles of Neutron Powder-Diffraction Peaks for Structure Refinement, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS .
- H. M. Rietveld, A Profile Refinement Method for Nuclear and Magnetic Structures, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS .
- B. H. Toby and R. B. Von Dreele, GSAS-II: The Genesis of a Modern Open-Source All Purpose Crystallography Software Package, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS .
- T. Kurobori, M. Liu and Y. Hirose, Temperature Dependence of the Near-IR Emission and Its Absorption in Oxygen-Doped BaFBr Crystals, Jpn. J. Appl. Phys., 2001, 40, L642–L645 CrossRef CAS .
- V. V. Mikhaylin and M. A. Terekhin, Luminescence Excitation in BaF2-BaCl2 and BaF2-BaBr2 Synchrotron Radiation, Rev. Sci. Instrum., 1989, 60, 2545–2547 CrossRef .
- V. V. Mikhaylin and M. A. Terekhin, Luminescence Excitation of Barium Fluorohalides by Using Synchrotron Radiation, Nucl. Instrum. Methods Phys. Res., Sect. A, 1989, 282, 607–609 CrossRef .
- Z. Liu, M. A. Stevens-Kalceff and H. Riesen, Effects of Postannealing on the Photoluminescence Properties of Coprecipitated Nanocrystalline BaFCl:Sm3+, J. Phys. Chem. A, 2013, 117, 1930–1934 Search PubMed .
- A. Sillen and Y. Engelborghs, The Correct Use of “Average” Fluorescence Parameters, Photochem. Photobiol., 1998, 67, 475–486 CrossRef CAS .
- W. Qin, K. Jang, S. Park, Y.-I. Lee and C. Kim, The Mechanism of Photobleaching in Sm2+-Doped Alkaline-Earth Fluorohalides, J. Lumin., 2005, 113, 9–16 CrossRef CAS .
- J. J. Schuyt and G. V. M. Williams, Oxygen-Impurity Charge Transfer in NaMgF3:Ln (Ln = Yb, Sm, or Eu): Establishing the Lanthanide Energy Levels in NaMgF3, J. Lumin., 2019, 211, 413–417 CrossRef CAS .
- J. C. Gâcon, J. C. Souillat, J. Seriot, F. Gaume-Mahn and B. Di Bartolo, Some Aspects of the Sm2+ Centre Relaxation in BaClF, J. Lumin., 1979, 18–19, 244–248 Search PubMed .
- R. Jaaniso and H. Bill, f–f and f–d Transition Interference in Sm2+:SrFCl, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 2389–2392 CrossRef CAS PubMed .
- Y. R. Shen and W. B. Holzapfel, Effect of Pressure on Energy Levels of Sm2+ in BaFCl and SrFCl, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 15752–15762 Search PubMed .
- Y. Shen and K. L. Bray, Effect of Pressure and Temperature on 4f–4f Luminescence Properties of Sm2+ ions in MFCl crystals (M = Ba, Sr, and Ca), Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 11944–11958 CrossRef CAS .
- P. Pal, T. Penhouet, V. D'Anna and H. Hagemann, Effect of Temperature and Pressure on Emission Lifetime of Sm2+ Ion Doped in MFX (M = Sr, Ba; X = Br, I) Crystals, J. Lumin., 2013, 142, 66–74 CrossRef CAS .
- J. Christmann and H. Hagemann, New Insights Into the Influence of the 4f55d1 State in the 4f6 Electronic Configuration of Sm2+ in Crystal Hosts, J. Phys. Chem. A, 2019, 123, 2881–2887 CrossRef CAS PubMed .
- P. Mikhail, K. Ramseyer, G. Frei, F. Budde and J. Hulliger, Bleaching of Sm2+ During Photoluminescence and Cathodoluminescence, Opt. Commun., 2001, 188, 111–117 Search PubMed .
- J. J. Schuyt and G. V. M. Williams, Quenching of the Sm2+ Luminescence in NaMgF3:Sm via Photothermal Ionization: Alternative Method to Determine Divalent Lanthanide Trap Depths, Appl. Phys. Lett., 2019, 115, 181104 CrossRef .
- Z.-y He, Y.-s Wang, S. Li and X.-r Xu, Dynamic Studies on the Time-Resolved Fluorescence of Sm2+ in BaCl2, J. Lumin., 2002, 97, 102–106 Search PubMed .
- J. F. Scott, Raman Spectra of BaClF, BaBrF, and SrClF, J. Chem. Phys., 1968, 49, 2766–2769 CrossRef CAS .
- L. Cui, Z. Dong, D. Yu, Y. Wang and A. Meijerink, High-Sensitivity Luminescent Temperature Sensors: MFX:1%Sm2+ (M = Sr, Ba, X = Cl, Br), Sci.
Adv., 2024, 10, eado7737 CrossRef CAS PubMed .
- K. Takahashi, K. Kohda, J. Miyahara, Y. Kanemitsu, K. Amitani and S. Shionoya, Mechanism of Photostimulated Luminescence in BaFX:Eu2+ (X = Cl,Br) Phosphors, J. Lumin., 1984, 31–32, 266–268 CrossRef CAS .
- T. Inerbaev, A. Akilbekov, D. Kenbayev, A. Dauletbekova, A. Shalaev, E. Polisadova, M. Konuhova, S. Piskunov and A. I. Popov, Color Centers in BaFBr Crystals: Experimental Study and Theoretical Modeling, Materials, 2024, 17, 3340 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
 .15,18–21 Negatively charged oxide defects are able to store holes,15,16,19 while positively charged halide vacancies may serve as electron traps (i.e., F color centers F(X−)). Although the involvement of color centers in the storage mechanism of photostimulable phosphors is well established,15,22–24 their role in the dynamics of electron trapping in photoexcitable phosphors remains an open question.4,5,8,25
.15,18–21 Negatively charged oxide defects are able to store holes,15,16,19 while positively charged halide vacancies may serve as electron traps (i.e., F color centers F(X−)). Although the involvement of color centers in the storage mechanism of photostimulable phosphors is well established,15,22–24 their role in the dynamics of electron trapping in photoexcitable phosphors remains an open question.4,5,8,25

 ), difference curve (
), difference curve ( , offset for clarity), and tick marks (
, offset for clarity), and tick marks ( ) corresponding to the calculated positions of the diffraction maxima are shown. (b) Representative TEM image showing nanoplates in two different orientations: base-lying and side-lying. (c) Size distribution histograms obtained after measuring 150 nanoplates.
) corresponding to the calculated positions of the diffraction maxima are shown. (b) Representative TEM image showing nanoplates in two different orientations: base-lying and side-lying. (c) Size distribution histograms obtained after measuring 150 nanoplates.



 . This reaction bleached Sm3+ centers that were initially luminescent. Photodissociation of oxide–halide ion vacancy pairs and diffusion of the resulting color centers is a key piece underlying the functionality of photostimulable BaFBr:Eu2+ phosphors.15,23,24 Diffusion of carrier traps was also brought up in studies of BaFCl:Sm3+,12 BaFCl:Tm3+,4 BaLiF3:Sm3+,6 and CaF2:Sm3+.8 It must be invoked here to rationalize the observation that the initial emission intensity from Sm3+ was not recovered after dark ionization following the first exposure to UVC radiation. In other words, the first exposure to UVC radiation caused an irreversible structural change that bleached Sm3+ centers that were initially luminescent. Thus, unlike the case of type I Sm3+, a dynamic structural component appeared to be involved in the initial photoreduction of type II Sm3+. Radiation-induced structural distortions were also observed in NaMgF3:Sm.45 Luminescent Sm2+ centers generated via diffusion of color centers underwent slow dark ionization to yield non-luminescent Sm3+ and regenerate distant O2− hole traps via long-distance tunneling (Sm2+ + O− → Sm3+ + O2−). For completeness we note that the bimodal distribution proposed here bears resemblance to that described by Eachus et al. for photostimulable centers in BaFBr:Eu2+,21 as both rely on a distribution of distances between carrier traps to rationalize experimental results.
. This reaction bleached Sm3+ centers that were initially luminescent. Photodissociation of oxide–halide ion vacancy pairs and diffusion of the resulting color centers is a key piece underlying the functionality of photostimulable BaFBr:Eu2+ phosphors.15,23,24 Diffusion of carrier traps was also brought up in studies of BaFCl:Sm3+,12 BaFCl:Tm3+,4 BaLiF3:Sm3+,6 and CaF2:Sm3+.8 It must be invoked here to rationalize the observation that the initial emission intensity from Sm3+ was not recovered after dark ionization following the first exposure to UVC radiation. In other words, the first exposure to UVC radiation caused an irreversible structural change that bleached Sm3+ centers that were initially luminescent. Thus, unlike the case of type I Sm3+, a dynamic structural component appeared to be involved in the initial photoreduction of type II Sm3+. Radiation-induced structural distortions were also observed in NaMgF3:Sm.45 Luminescent Sm2+ centers generated via diffusion of color centers underwent slow dark ionization to yield non-luminescent Sm3+ and regenerate distant O2− hole traps via long-distance tunneling (Sm2+ + O− → Sm3+ + O2−). For completeness we note that the bimodal distribution proposed here bears resemblance to that described by Eachus et al. for photostimulable centers in BaFBr:Eu2+,21 as both rely on a distribution of distances between carrier traps to rationalize experimental results.





