Pursuing red thermally activated delayed fluorescence upon increasing the degree of branching in phenothiazine-trithienyltriazine push–pull compounds
Received
23rd August 2025
, Accepted 10th November 2025
First published on 11th November 2025
Abstract
In this study, three new push–pull compounds bearing phenothiazine as the electron donor and trithienyltriazine as the electron acceptor connected by triple bond π-bridges and arranged in dipolar (TRZ1), quadrupolar (TRZ2) and octupolar (TRZ3) molecular structures were designed and synthesized. The efficiencies and rates of their excited state deactivation pathways, strongly modulated by the environment, were unveiled through advanced time resolved spectroscopies with nanosecond and femtosecond temporal resolution. Highly efficient fluorescence and intersystem crossing were found to occur in non polar solvents, which justify all the absorbed quanta. Emission measurements in a non polar rigid matrix at low temperature uncovered small singlet-to-triplet energy gaps for these molecules. Indeed, in fairly polar media, an intramolecular charge transfer state quasi-isoenergetic to the triplet excited state was stabilized and populated, resulting in reverse intersystem crossing followed by orange/red delayed fluorescence. Similarly, delayed fluorescence was clearly detected in thin films of TRZ1–3 at room temperature. Upon increasing the degree of branching among the molecules in this series, the emission color in the solid state was turned from orange to deep red and the amplitude of the delayed component was largely enhanced. The mechanistic reason underlying this behavior could be found in the relatively faster intersystem crossing than prompt fluorescence in the multi-branched compounds. Our results demonstrate the positive role played by the degree of branching in boosting red delayed fluorescence in all-organic materials for applications in third generation organic light emitting diodes.
Introduction
Thermally activated delayed fluorescence (TADF) is an intriguing emissive phenomenon exploited in third and fourth generation organic light emitting diodes (OLEDs).1–6 OLEDs have attracted great interest for display technologies in comparison to traditional liquid crystals for their unique features such as fast response time, thinness, flexibility, transparency, no backlighting, wide viewing angle and low power lighting.7 Over the past few decades, luminescent materials for OLED devices have undergone development from organic fluorophores (first generation OLEDs based on fluorescence) to organometallic phosphors (second generation OLEDs based on phosphorescence), with internal quantum efficiencies (IQEs) increasing from 25% to 100%. On the other hand, TADF allows to harvest triplet excitons to give delayed fluorescence emission following the reverse intersystem crossing from the triplet to the singlet excited state.8 TADF-based OLEDs combine the advantage of low cost all-organic fluorescent materials with the high-efficiency of phosphorescent OLEDs.9 While many examples of blue, green and yellow TADF emitters have been reported in the recent literature, developing effective orange and red TADF fluorophores remains a challenge.10–18 This is mainly due to the energy gap law: longer emission wavelengths imply a narrower S1–S0 energy gap, with the consequent prevalence of nonradiative decay processes. In order to boost the efficiency of red TADF emitters, ad hoc molecular design strategies have been developed. Push–pull systems exhibiting strong intramolecular donor (D)–acceptor (A) interactions have been considered to obtain a red-shifted emission and achieve the small singlet-to-triplet energy gap necessary to enable the reverse intersystem crossing process.19,20 The goal of a significant fluorescence efficiency has been generally pursued by increasing the molecular conjugation of the considered organic molecules.21,22
Our group has recently contributed to the development of new red TADF emitters by synthetizing and characterizing new phenothiazine-naphthalimide derivatives, where the phenothiazine is the electron donor and the naphthalimide the electron acceptor portion.22,23 In this investigation, we consider a stronger electron acceptor unit, constituted by a triazine derivative. Triazine is indeed a well know structural motif as a remarkable electron acceptor in TADF emitters.24–30 Previous literature work has described the photophysics of phenothiazine-triphenyltriazine conjugates bearing single bond linkers, highlighting the occurrence of intense yellow TADF.17,28,29,31–33 In the present work, with the aim being increasing the molecular conjugation to enable efficient orange-red TADF, we synthetized phenothiazine-triazine derivatives bearing triple bonds as π-bridges as well as arranged in dipolar (TRZ1), quadrupolar (TRZ2) and octupolar (TRZ3) structures characterized by different degrees of branching (Chart 1).34 Moreover, in the molecular structures here considered, the central triphenyltriazine unit was replaced with a trithienyltriazine since that employing thiophenes, instead of phenyls, has been sometimes described in the literature to imply a red shift in the absorption and emission spectra.35 Here, a comprehensive photophysical study was carried out for the new TRZ1–3 chromophores, both in solution and in solid state thin films, utilizing state-of-the-art time resolved spectroscopies, such as nanosecond and femtosecond transient absorption and femtosecond broadband fluorescence up conversion. A deep insight was gained into the yields, rates and energetics of the fluorescence and intersystem crossing processes36,37 of such push–pull compounds, which may inspire future ideas for new optimized red TADF materials for third generation OLEDs based on chemical principles rather than trial-and-error approaches.
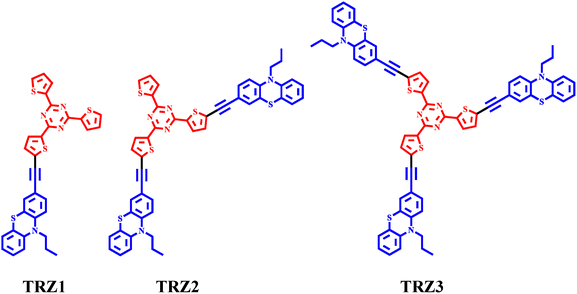 |
| | Chart 1 Molecular structures of the investigated compounds. | |
Results and discussion
Synthesis and characterization
The synthetic procedure for the TRZ1–3 derivatives is summarized in Scheme 1 and detailed in the SI. These compounds were synthesized via Sonogashira cross-coupling reaction between Mono-TRZ-Th, Di-TRZ-Th, and Tri-TRZ-Th with PTZ4 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of triethylamine and tetrahydrofuran. The reaction was performed at 60 °C in the presence of Pd(PPh3)2Cl2 and CuI as catalysts, resulting in TRZ1–3 in 69%, 57%, and 67% yield, respectively.
:1 mixture of triethylamine and tetrahydrofuran. The reaction was performed at 60 °C in the presence of Pd(PPh3)2Cl2 and CuI as catalysts, resulting in TRZ1–3 in 69%, 57%, and 67% yield, respectively.
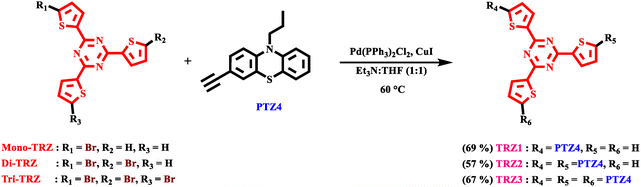 |
| | Scheme 1 Synthetic route for triazine-based chromophores TRZ1–3. | |
All the compounds were purified by column chromatography using hexane:dichloromethane as the eluent and were well-characterized by 1H NMR spectroscopy, 13C NMR spectroscopy, and HRMS techniques (see Fig. S1–S23).
Absorption and emission properties
Fig. 1 shows the absorption and emission spectra of TRZ1–3 in a non polar solvent, such as cyclohexane. The molecular structure effect on the spectra is small and implies a slight red shift of both the absorption and emission bands upon passing from the mono-branched to the double-branched and tri-branched derivatives. On the other hand, the fluorescence properties (Table 1) were significantly affected by the molecular structure with a fluorescence quantum yield (ΦF) of 18% for TRZ1, 26% for TRZ2 and 35% for TRZ3 in cyclohexane. Similarly, the fluorescence lifetime (τF) was enhanced on going from TRZ1 (0.8 ns) to TRZ2 (1.0 ns) and TRZ3 (1.2 ns). As a result, the fluorescence rate constant, computed as kF = ΦF/τF, was found to be slightly larger in TRZ3 (2.9 × 108 s−1) relative to TRZ2 (2.6 × 108 s−1) and TRZ1 (2.2 × 108 s−1).
 |
| | Fig. 1 Normalized Absorption (upper graphs) and Emission (lower graphs) spectra of TRZ1–3 in cyclohexane (left) and of TRZ1 in solvents of different polarities (right). To record the emission spectra for each sample, excitation was carried out at the corresponding absorption maximum above 400 nm. Image: Fluorescence of TRZ1 in different solvents under UV light at 365 nm. | |
Table 1 Fluorescence properties of TRZ1–3 in cyclohexane (upper table) and of TRZ1 in solvents of different polarities (lower table)
| Structure effect in cyclohexane |
|
|
Φ
F
|
τ
F (ns) |
k
F (s−1) |
|
TRZ1
|
0.18 |
0.8 |
2.2 × 108 |
|
TRZ2
|
0.26 |
1.0 |
2.6 × 108 |
|
TRZ3
|
0.35 |
1.2 |
2.9 × 108 |
| Solvent effect for TRZ1 |
|
|
ε
|
Φ
F
|
τ
F (ns) |
k
F (s−1) |
|
n-Hexane |
1.89 |
0.13 |
0.8 |
1.6 × 108 |
| Cyclohexane |
2.02 |
0.18 |
0.8 |
2.2 × 108 |
| Toluene |
2.38 |
0.63 |
3.3 |
1.9 × 108 |
| Anisole |
4.33 |
0.23 |
2.5 |
9.2 × 107 |
| THF |
7.58 |
0.07 |
2.0 |
3.5 × 107 |
| MeCN |
36.6 |
0.001 |
— |
— |
The effect of the solvent on the absorption and fluorescence spectra of TRZ1 is shown in Fig. 1 and detailed in Fig. S26 and S27 for TRZ2 and TRZ3. The absorption spectra undergo a small shift, mainly determined by the solvent polarizability. A very significant red shift is instead observed for the fluorescence spectrum with the solvent dielectric constant.38,39 The fluorescence spectrum shows an important vibrational structure in the less polar solvents (n-hexane and cyclohexane) while becoming a broad structureless band in the relatively more polar toluene, anisole and tetrahydrofuran solvents. This strong positive fluorosolvatochromism implies a significant change in the colour of the emission with the solvent: from the blue fluorescence in n-hexane and cyclohexane, to yellow in toluene, orange in a toluene/anisole mixture and red in anisole and tetrahydrofuran (Fig. 1). No significant visible emission was observed in the more polar acetonitrile and dimethylsulfoxide solvents under ultraviolet light. For TRZ1–3, the fluorescence spectra recorded in most solvents show of a single emission band (Fig. 1), no significant excitation wavelength effect was observed on the emission spectra (Fig. S29) and the excitation spectra were found to overlap well the relative absorption spectrum (Fig. S25 and S29). The only evidence for a small contribution of a second emitting species (possibly a conformer, as discussed in literature studies about phenothiazine)29,40 was found while investigating the fluorescence spectra in tetrahydrofuran (Fig. S30). The fluorescence quantum yield was largely affected by the solvent polarity, with similar trends also observed for the fluorescence lifetime and rate constant of TRZ1–3 (Table 1 and Tables S2–S4). The fluorescence quantum yield was found to increase from 0.13 in n-hexane to 0.18 in cyclohexane and 0.63 in toluene for TRZ1. Similar fluorescence quantum yields (0.63), lifetimes (3.3 ns) and rate constants (1.9 × 108 s−1) were measured for all the TRZ1–3 investigated compounds in toluene. This finding suggests that excited state symmetry breaking and localization of the excitation on a single branch of the molecular structure take place for the quadrupolar and octupolar derivatives.41–43 Upon further increasing the solvent dielectric constant, a significant fluorescence quenching was observed in anisole (ΦF = 0.23 for TRZ1) and tetrahydrofuran (ΦF = 0.07 for TRZ1). This double trend of the fluorescence properties with the solvent dielectric constant indicates the presence of two competitive deactivation pathways to the fluorescence, one operative in non polar media (such as n-hexane and cyclohexane) and one operative in polar solvents (anisole and tetrahydrofuran). In particular, in the most polar solvent investigated in detail (tetrahydrofuran) the photoinduced intramolecular charge transfer competitive pathway seems to be more efficient leading to a more effective fluorescence quenching for the octupolar TRZ3 (ΦF = 0.02) and for the quadrupolar TRZ2 (ΦF = 0.03) than for the dipolar TRZ1 (ΦF = 0.07).44
Quantum chemical calculations at the DFT level performed on the ground state optimized geometry showed that the S0 → S1 transition is mainly described by the HOMO–LUMO configuration for TRZ1–3 (Fig. S31 and Tables S5–S8). The electron density of the HOMOs for TRZ1–3 is displaced over the one/three phenothiazine units and the ethynyl thiophene spacers. As for the LUMOs the electron density is mainly localized on the central thienyl triazine core. The absorption band at longest wavelength are thus characterized by a significant intramolecular charge transfer character. The computationally predicted absorption spectra exhibited a remarkable agreement with the experimental ones (Fig. S32).
The redox potentials of TRZ1–3 were evaluated by cyclic voltammetry and differential pulse voltammetry (Fig. S33, S34 and Table S9). All the measurements were performed in dry dichloromethane at room temperature using 0.1 M tetrabutylammonium hexafluorophosphate as a supporting electrolyte. The electrochemical behaviour of TRZ1–3 reveals both oxidation and reduction processes, arising from the incorporation of donor and acceptor units within their molecular structures. All the three derivatives exhibit two oxidation waves in the anodic region and two reduction waves in the cathodic region in their cyclic voltammograms. The oxidation waves observed at lower potentials are attributed to the phenothiazine unit,39 whereas the oxidation wave at higher potentials corresponds to the thiophene spacer.45 The reduction waves for TRZ1–3 in the cathodic region are due to the triazine moiety.46
Ultrafast spectroscopy
The nature of these non radiative deactivation pathways was unveiled through the investigation of the ultrafast excited state dynamics via femtosecond transient absorption and broadband fluorescence up conversion measurements for TRZ1–3 in selected solvents of different polarities (cyclohexane, toluene and tetrahydrofuran).
The results of the femtosecond transient absorption experiments for TRZ1 in cyclohexane and tetrahydrofuran are shown in Fig. 2 as representative examples. The transient absorption spectra in cyclohexane at early delays exhibit a broad positive excited state absorption (ESA) band peaked at 575 nm and 710 nm as well as a negative signal due to stimulated emission (SE) peaked around 500 nm. While this transient spectrum decays, a positive broad ESA peaked below 500 nm rises and remains also at long delays after photoexcitation. The fitting of these data revealed the presence of four exponential components (Table 2 and Fig. S35): a first component with a lifetime of 1.6 ps assigned to vibrational cooling (VC), a second component with a lifetime of 240 ps assigned to structural relaxation (SR), a third component with a lifetime of 800 ps in line with the fluorescence decay assigned to the lowest excited singlet state (S1), a fourth component with an Infinite lifetime as not decaying in the investigated time window of ca. 3 ns assigned to the lowest excited triplet state (T1). Such assignments were confirmed by the fluorescence up conversion experiments (Fig. 3 and Fig. S44–S46 as well as Table 2). In cyclohexane, the time resolved emission spectra highlighted a structured fluorescence centered around 525 nm, formed at early delays after excitation and decaying within the investigated time window of 3 ns. Such emission was assigned to the locally excited singlet state populated upon light absorption (S1,LE). The ultrafast experiments in cyclohexane thus undisclosed the occurrence of intersystem crossing for all the TRZ1–3 compounds, which takes place in 800 ps for TRZ1, 1000 ps for TRZ2 and 1200 ps for TRZ3 (see Table 2 and Fig. S35–S37).
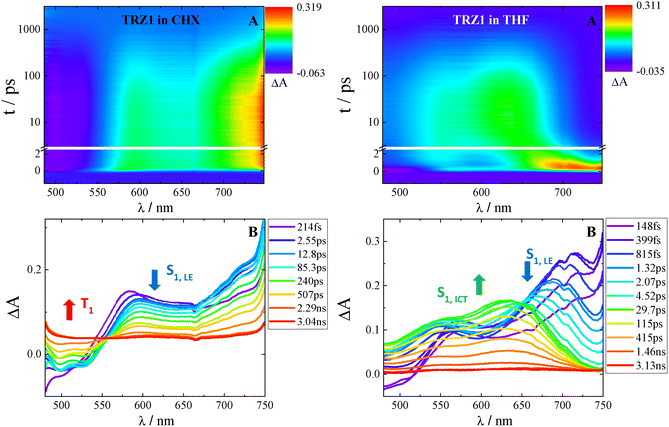 |
| | Fig. 2 Femtosecond transient absorption of TRZ1 in cyclohexane (left) and tetrahydrofuran (right): (A) tridimensional data matrix ΔA as a function of wavelength and time; (B) time resolved absorption spectra at different delays after photoexcitation. | |
Table 2 Lifetimes resulting from the global analysis of the femtosecond transient absorption (TA) and fluorescence up conversion (FUC) data for TRZ1–3 in cyclohexane (CHX) and tetrahydrofuran (THF)
 |
| | Fig. 3 Femtosecond broadband fluorescence up conversion of TRZ1 in cyclohexane (left) and tetrahydrofuran (right): (A) tridimensional data matrix fluorescence intensity as a function of wavelength and time; (B) time resolved emission spectra at different delays after photoexcitation. | |
When TRZ1 was investigated in the more polar tetrahydrofuran solvent through femtosecond transient absorption (Fig. 2), the initial spectrum characterized by the 550 nm and 710 nm ESA and by the 500 nm SE was found to evolve in time toward another species, not observed in cyclohexane, characterized by an absorption spectrum with broad bands at 530 nm and 630 nm. This transient was then found to decay within the investigated time window. The global fit revealed the presence of five exponential components for TRZ1 in tetrahydrofuran (Table 2 and Fig. S41): the first two of 0.52 ps and 1.4 ps compatible with solvent relaxation (solv.)47 and with a spectral shape resembling the S1,LE state, the third component of 300 ps associated to structural relaxation (SR) possibly among different conformers, the forth component of few nanoseconds assigned to an intramolecular charge transfer state (S1,ICT) stabilized in this polar solvent, a residual Inf component relative to a still sizable population of the T1 state. These results were confirmed by the fluorescence up conversion experiments in tetrahydrofuran (Fig. 3 and Fig. S50–S52). The emission spectra temporal dynamics appeared to be completely different from that in the non polar cyclohexane. The spectrum at early delays is a vibrationally structured band at 525 nm, resembling the S1,LE emission. This spectrum then undergoes an important red shift in time. The final emission is constituted by a broad band centered at ca. 650 nm and assigned to the S1,ICT in a polar solvent such as tetrahydrofuran.48 The ultrafast experiments in tetrahydrofuran for TRZ1, but also for TRZ2 and TRZ3, unveil the occurrence of intramolecular charge transfer which is, besides intersystem crossing, the other competitive pathway to the fluorescence operative for these molecules in polar solvents. The ultrafast data revealed a S1,ICT lifetime of 2.0 ns for TRZ1 while being 1.4 ns for both TRZ2 and TRZ3 in tetrahydrofuran (see Table 2). These findings suggest a faster non radiative deactivation of the ICT excited state to the ground state for the more complex quadrupolar and octupolar structures relative to the dipolar system, in agreement with their smaller fluorescence quantum yields in this solvent.44
Triplet properties
The intersystem crossing pathway of triplet formation was investigated in more detail for TRZ1–3 through nanosecond transient absorption experiments.
The nanosecond transient spectra recorded in cyclohexane (Fig. 4 and Fig. S53) exhibited a region of negative signal due to ground state bleaching (GSB) centered around 400 nm and a broad band of positive ESA peaked at ca. 480 nm, with the maximum slightly red shifting upon increasing conjugation in the molecular structure. The ESA peak resulted somehow red shifted (500–520 nm) also when the experiments were carried out in more polar solvents (Fig. S54–S56). The observed transient species was found to decay in hundreds of nanoseconds in air equilibrated and in tens of microseconds in nitrogen purged solution, revealing an exceptional sensitivity to the presence of molecular oxygen (Fig. S57). This transient was found to be successfully sensitized by a high energy triplet donor, such as 2,2′-dithienyl ketone (DTK).49 These findings proved unambiguously that the detected excited species was the lowest excited triplet state (T1) of these molecules. Sensitization experiments also allowed to obtain information about the triplet extinction coefficients, found to be 14400 M−1 cm−1 for TRZ1, 17450 M−1 cm−1 for TRZ2 and 18280 M−1 cm−1 for TRZ3, slightly enhanced upon increasing molecular conjugation. Relative actinometry measurements allowed to get an accurate evaluation of the triplet quantum yield (ΦT), which was revealed to be 92% for TRZ1, 58% for TRZ2 and 36% for TRZ3 in cyclohexane (see Table 3). The triplet yields were observed to significantly decrease upon increasing the solvent dielectric constant (Table S11). While the main decay pathway is the intersystem crossing in cyclohexane (e.g. for TRZ1ΦF = 18% and ΦT = 92%), a relatively more important fluorescence is observed in toluene (e.g. for TRZ1ΦF = 63% and ΦT = 22%). In tetrahydrofuran, both the intersystem crossing and the fluorescence are revealed to be low efficient processes (e.g. for TRZ1ΦF = 7% and ΦT = 5%), pointing to the crucial role played by the intramolecular charge transfer in polar media. Deactivation of the low energetic S1,ICT will likely take place mainly through internal conversion to the ground state for these molecules.
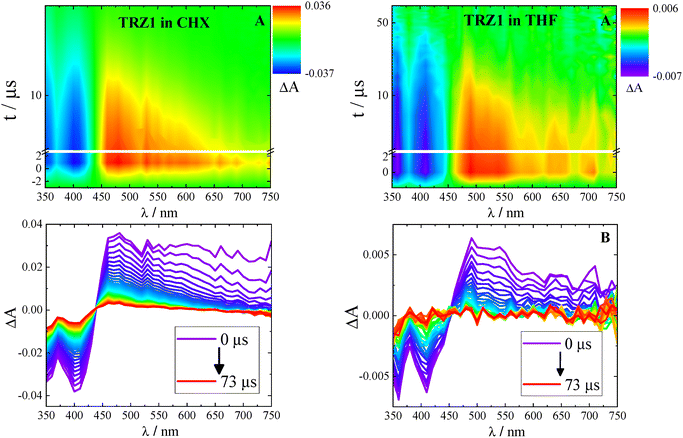 |
| | Fig. 4 Nanosecond transient absorption results: (A) tridimensional data matrix ΔA as a function of wavelength and time; (B) time resolved absorption spectra at different delays after photoexcitation. Left: TRZ1 in cyclohexane. Right: TRZ1 in tetrahydrofuran. | |
Table 3 Triplet properties of TRZ1–3 in cyclohexane (upper table); fluorescence and triplet quantum yields for TRZ1 in solvents of different polarities (lower table)
| Structure effect in cyclohexane |
| Compound |
Solvent |
τ
T,air (ns) |
τ
T,N2 (µs) |
ε
T (M−1 cm−1) |
Φ
T
|
|
TRZ1
|
CHX |
200 |
25 |
14400 |
0.92 |
|
TRZ2
|
180 |
42 |
17450 |
0.58 |
|
TRZ3
|
156 |
40 |
18280 |
0.36 |
| Solvent effect for TRZ1 |
| Compound |
Solvent |
Φ
F
|
Φ
T
|
|
TRZ1
|
CHX |
0.18 |
0.92 |
| Tol |
0.63 |
0.22 |
| THF |
0.07 |
0.05 |
Delayed fluorescence
Information about the singlet and triplet excited state energies was gained through emission measurements at 77 K. Steady and delayed emission spectra were recorded, which should correspond to the fluorescence and phosphorescence spectra under these experimental conditions (Fig. 5 and Fig. S58, S59). Phosphorescence spectra were found to be red shifted relative to the fluorescence and peaked at ca. 600 nm for TRZ1 and ca. 650 nm for TRZ2–3. The shoulder observed in the delayed spectrum of TRZ2–3 around 560 nm may be due to some delayed fluorescence following triplet–triplet annihilation, as also reported in literature studies.8 From the onset of the fluorescence and phosphorescence spectra, the singlet and triplet excited state energies were evaluated, respectively (see Fig. 5 left). Both the singlet and triplet excited state energies were found to decrease upon increasing the molecular branching on going from TRZ1 to TRZ2 to TRZ3 (Table 4). The singlet-to-triplet energy gaps (ΔEST) were found to be in the range 0.23–0.38 eV, compatible with the occurrence of thermally activated delayed fluorescence.50
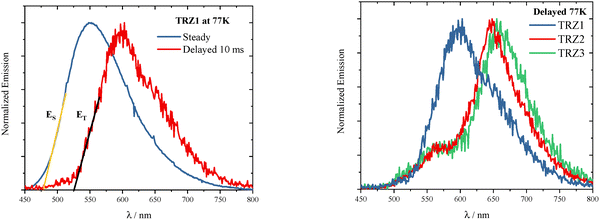 |
| | Fig. 5 Emission spectra recorded in MethylCyclohexane/3-MethylPentane mixture at 77 K. Left: Steady and Delayed emission spectra for TRZ1 and determination of the singlet and triplet excited state energies. Right: Delayed emission spectra for TRZ1–3. | |
Table 4 Singlet (ES) and triplet (ET) excited state energies as derived from the steady and delayed emission spectra recorded at 77K (given in nm and in eV) and relative singlet-to-triplet energy gaps (ΔEST in eV) for TRZ1–3
| Compound |
E
S (nm eV−1) |
E
T (nm eV−1) |
ΔEST (eV) |
|
TRZ1
|
479/2.59 |
525/2.36 |
0.23 |
|
TRZ2
|
489/2.54 |
575/2.16 |
0.38 |
|
TRZ3
|
504/2.46 |
591/2.10 |
0.36 |
The significant intersystem crossing observed for these all-organic compounds and the rather small ΔEST pushed us to investigate further their delayed fluorescence ability. Therefore, fluorescence decay kinetics were recorded on long time scales of 1–2 µs through the Time correlated single photon counting (TC-SPC) technique. In rather polar solvents, such as tetrahydrofuran, a clear biexponential decay was observed (Fig. 6 left and Fig. S61), with a lifetime of few nanoseconds assigned to the prompt fluorescence also detected in a shorter time window (see Fig. S28) as well as a lifetime of 350–370 ns (Table S13). This longer lifetime was found to be consistent with the triplet lifetime measured via nanosecond laser flash photolysis in air equilibrated solutions (Table S11). The amplitude of this long lived component in describing the fluorescence decay was found to increase on going from TRZ1 (0.01%) and TRZ2 (2%) to TRZ3 (26%, see Table S13). Interestingly, in the case of TRZ1, for which a relatively smaller energy gap and a blue shifted phosphorescence was found, such long lived emission was observed also in solvents of lower polarity, such as toluene and anisole (Fig. S61). The long lived emission detected for TRZ1–3 at room temperature through these experiments was assigned to delayed fluorescence occurring from the singlet excited state following reverse intersystem crossing from T1. Given the small but still sizable ΔEST measured in the low polar methylcyclohexane/3-methylpentane mixture between the S1,LE and T1 for these compounds, it is likely that the zero gap condition favouring reverse intersystem crossing and delayed fluorescence is achieved at room temperature between the S1,ICT stabilized in more polar solvents (such as anisole and tetrahydrofuran) and the T1,51 as sketched in Fig. 7. Indeed, in literature studies, it has been found that polar solvents tend to stabilize S1 states, while the receiver triplet is much less sensitive to solvent effects.23,52–54 By comparing the highly solvatochromic fluorescence and the phosphorescence spectra recorded for TRZ2–3, it can be inferred that this energy match is achieved in tetrahydrofuran (Fig. S60). Given the blue shifted phosphorescence measured for TRZ1, it is feasible that the reverse intersystem crossing becomes energetically possible even in solvents of lower polarity (e.g. toluene or toluene/anisole) in this case (Fig. S60). In Fig. 7, histograms giving the detailed rate constant values of prompt fluorescence (kPF = ϕF/τS1) and intersystem crossing (kISC = ϕT/τS1) for TRZ1–3 in tetrahydrofuran, where an apparent delayed fluorescence is observed, are also reported. It is noteworthy that the computed kPF and kISC are of the same order of magnitude for all the three compounds. Interestingly, kPF is larger than kISC for TRZ1 ((kPF = 3.5 × 107 s−1 and kISC = 2.5 × 107 s−1), kPF is equal to kISC for TRZ2 ((kPF = 2.1 × 107 s−1 and kISC = 2.1 × 107 s−1) and kPF is lower than kISC for TRZ3 (kPF = 1.4 × 107 s−1 and kISC = 3.6 × 107 s−1). The larger kPF for TRZ1 compared to TRZ2 and TRZ3 in tetrahydrofuran may be due to its relatively lower excited state intramolecular charge transfer degree. These data give a justification to why, under these experimental conditions of roughly zero ΔEST in tetrahydrofuran, a more important delayed fluorescence is observed for TRZ3 than for TRZ2 than for TRZ1.55,56 A method described in the literature by Monkman et al.8 was employed to obtain an estimate of the reverse intersystem crossing rate constant (krISC) for the investigated compounds. The employed method and the obtained results are reported in detail in Table S14. The obtained krISC values are quite low but show a trend of increasing upon increasing the degree of branching and suggest that TRZ2 and TRZ3 may be considered TADF materials (krISC = 1.1 × 105 and 5.9 × 105 s−1, respectively).
 |
| | Fig. 6 Fluorescence decay kinetics recorded on a long time window (1–2 µs) through the time correlated single photon counting technique for TRZ1–3 in tetrahydrofuran (THF) at room temperature (left) and for TRZ1 in toluene at different temperatures in the 77–343 K range (right). | |
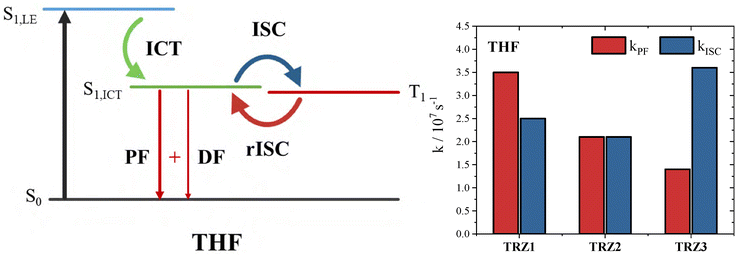 |
| | Fig. 7 Left: Sketch of the excited state deactivation for the investigated compounds in tetrahydrofuran (THF). Right: Histogram detailing the prompt fluorescence (kPF) and intersystem crossing (kISC) rate constants for TRZ1–3 in tetrahydrofuran. | |
The effect of temperature on the fluorescence decay kinetics in toluene was investigated to gain information about the thermally activated delayed fluorescence behaviour of these molecules.57 At 77 K, in a rigid frozen toluene matrix, the fluorescence kinetics shows a clear biexponential decay for all the TRZ1–3 with the prompt fluorescence exhibiting lifetimes of few nanoseconds and a delayed fluorescence component in the hundreds of nanoseconds timescale (Fig. S62 and Table S15). For TRZ1 (Fig. 6), the importance of such long lived component was found to be large and slightly enhanced upon raising the temperature up until 200 K, pointing to the thermal activation of the delayed fluorescence behaviour. At higher temperatures, when the toluene matrix begins to melt becoming a liquid environment, the delayed fluorescence was found to be less important (250 K) and then disappear (above room temperature), being possibly turned off by the competition of non radiative decay pathways in this case. When the other chromophores are considered (Fig. S62 and Table S15), the activation of the delayed fluorescence is apparent only between 77 K and 150 K (for TRZ2) and only between 77 K and 100 K (for TRZ3). This suggests that the competition of non radiative decay pathways in liquid solution may play a more crucial role in such more complex and flexible quadrupolar and octupolar structures compared to TRZ1.
Long lived emission in thin film
The three investigated molecules were also characterized for their absorption and emission properties in the solid state, in neat thin films prepared by spin coating. The absorption spectra of the films were found to be red shifted with respect to the solution spectra (Fig. S64). Moreover, both the absorption and the emission bands were found to be red shifted when passing from TRZ1 to TRZ2 to TRZ3, in line with the increase of branching in this series of molecules (Fig. 7 and Fig. S63). Emission in the neat films was found to be quite intense (quantum yields in Table S16) and centered at 605 nm for TRZ1, 620 nm for TRZ2 and 650 nm for TRZ3 (Fig. 8). This results in a bright orange to red emission for the thin films observed under ultraviolet light (Fig. 8). Interestingly, nanosecond transient absorption revealed a significant signal decaying with a lifetime of ca. 260 ns and characterized by a spectrum showing ground state bleaching around 430 nm and excited state absorption around 540 nm, both slightly red shifted relative to the solution spectrum (Fig. S67). Such kinetic and spectral features suggest that a significant triplet production occurs also in the solid state for TRZ1. The remarkable matching between the spectral position of the steady-state fluorescence of the films of TRZ1–3 and their phosphorescence at low temperature (Fig. S68) pushed us to go deeper into their possible delayed fluorescence capability. Indeed, the fluorescence kinetics of the films were found to show a biexponential decay, with the long component showing a lifetime of 350–400 ns slightly dependent on the molecular structure (Table S17). It is noteworthy that the weight of the delayed fluorescence was found to increase upon passing from TRZ1 (1.5%) to TRZ2 (21%) to TRZ3 (46%) and thus upon increasing the number of branches in the molecular structure of these triazine-phenothiazine fluorophores. The observation of a bright orange-red delayed fluorescence in such novel organic materials in the solid state is highly promising for their possible application in third generation OLED devices.
 |
| | Fig. 8 Normalized fluorescence spectra (upper left graph) and fluorescence decay kinetics on a long time window (upper right graph) for TRZ1–3 in thin film. Lower image: Fluorescence of TRZ1–3 films under UV light. | |
Further investigation of these films by using a micropulsed lamp and detecting their time resolved emission spectra (TRES) in the microsecond time scale allowed to observe, for the case of TRZ1, a long lasting emission characterized by lifetimes of ca. 50 µs (see Fig. S70), while the kinetics recorded for the TRZ2 and TRZ3 films roughly overlap the instrumental response function. For TRZ1, for which a less efficient delayed fluorescence relative to the other investigated fluorophores takes place according to the single photon counting kinetics, the long lived emission observed in film with the TRES analysis matches the phosphorescence centered around 600 nm detected at low temperature (Fig. S69). These findings point to the contribution of room temperature phosphorescence (RTP) in this solid state material on long time windows,33,38,58,59 which makes its potential for optoelectronic applications high.
Conclusions
In this study, three new push–pull compounds were designed and synthetized bearing trithienyltriazine as the electron acceptor and phenothiazine as the electron donor units, connected by triple bond π-bridges and arranged in dipolar (TRZ1), quadrupolar (TRZ2) and octupolar (TRZ3) structures. The goal of the synthetic effort has been to achieve strong donor–acceptor interactions and large conjugation in these molecules (through the π-linkers and upon increasing the degree of branching) to enable efficient long wavelength delayed fluorescence in these all-organic materials.
The dynamics of fluorescence and intersystem crossing were unveiled for these fluorophores to be strongly modulated by the environment through femtosecond and nanosecond transient absorption as well as femtosecond fluorescence up conversion experiments. Highly efficient fluorescence and intersystem crossing were revealed to be operative in non polar solvents. Fairly small singlet-to-triplet energy gaps (0.23–0.38 eV) were found through emission measurements in rigid matrix at low temperature. In fairly polar solvents, such as anisole or tetrahydrofuran, a stabilized intramolecular charge transfer state is populated, which becomes isoenergetic to the triplet excited state. Consequently, orange/red delayed fluorescence was clearly observed for TRZ1–3 at room temperature in such media. The relative rates of prompt fluorescence and intersystem crossing for the three molecules point to a relatively faster intersystem crossing than prompt fluorescence for the octupolar compound compared to the quadrupolar and dipolar molecules, justifying the larger amplitude of the delayed fluorescence observed for the three-branched molecule. Interestingly, orange emission was also detected for TRZ1 thin films. A more important and red delayed fluorescence was revealed for the two branched (TRZ2) and three branched (TRZ3) derivatives in the solid state, demonstrating the positive impact of increasing the degree of branching on boosting the red delayed fluorescence of these new organic materials. On the other hand, a long lived emission, which could be ascribed to room temperature phosphorescence, was detected for the dipolar TRZ1 molecule in the solid state. Our results suggest that these new all-organic materials are highly promising for harvesting triplet excitons in optoelectronic light emitting devices to give either bright orange room temperature phosphorescence or red thermally activated delayed fluorescence, depending on the degree of branching of their molecular structure.
Author contributions
P. M., C. M. and K. Y.: data curation, investigation, methodology, writing – original draft, writing – review & editing; E. L.: data curation, investigation, writing – review & editing; R. M.: conceptualization, supervision, writing – review & editing; B. C.: conceptualization, investigation, supervision, writing – original draft, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: Details about the synthesis and characterization (NMR and HRMS) of the compounds; the absorption and emission properties; the quantum chemical calculations; the electrochemical properties; the ultrafast spectroscopy and laser flash photolysis results; the temperature effect study; the thin film characterization. See DOI: https://doi.org/10.1039/d5tc03179f.
Acknowledgements
This work was financially supported by the European Union - NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY and by the MUR under the PRIN 2022 program (grant no. 2022RRFJC4). R. M. acknowledges the Science and Engineering Research Board (SERB) Project No. CRG/2022/000023 and STR/2022/000001, the Council of Scientific and Industrial Research (Project No. 01/3112/23/EMR-II) and STR/2022/000001, New Delhi and the Department of Science and Technology DST-EMR Project No. DST-EMR: EMR/2017/001637.We are grateful to the DST-FIST grant for the 500 MHz NMR facility and the Sophisticated Instrumentation Centre (SIC), Indian Institute of Technology (IIT) Indore. K. Y. thanks the University Grant Commission (UGC)-New Delhi for the fellowship.
Notes and references
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 1–20 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- L. Ge, W. Zhang, Y.-H. Hao, M. Li, Y. Liu, M. Zhou and L.-S. Cui, J. Am. Chem. Soc., 2024, 146, 32826–32836 CrossRef CAS PubMed.
- S. Kothavale, W. J. Chung and J. Y. Lee, J. Mater. Chem. C, 2021, 9, 528–536 RSC.
- Y. Xiao, H. Wang, Z. Xie, M. Shen, R. Huang, Y. Miao, G. Liu, T. Yu and W. Huang, Chem. Sci., 2022, 13, 8906–8923 RSC.
- Z. Cai, X. Wu, H. Liu, J. Guo, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 23635–23640 CrossRef CAS PubMed.
- J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 6, 1800255 CrossRef.
- X. Zhang, X. Liu, M. Taddei, L. Bussotti, I. Kurganskii, M. Li, X. Jiang, L. Xing, S. Ji, Y. Huo, J. Zhao, M. Di Donato, Y. Wan, Z. Zhao and M. V. Fedin, Chem. – Eur. J., 2022, 28, e202200510 CrossRef CAS PubMed.
- G. Tang, A. A. Sukhanov, J. Zhao, W. Yang, Z. Wang, Q. Liu, V. K. Voronkova, M. Di Donato, D. Escudero and D. Jacquemin, J. Phys. Chem. C, 2019, 123, 30171–30186 CrossRef CAS.
- L. An, R. Su, Y. Sun, C. Song and Q. Wang, Comput. Theor. Chem., 2023, 1228, 114277 CrossRef CAS.
- C. Montanari, M. Sheokand, A. Cesaretti, E. Calzoni, R. Misra and B. Carlotti, J. Phys. Chem. C, 2024, 128, 19688–19700 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766–3771 CrossRef CAS.
- E. Duda, D. Hall, S. Bagnich, C. L. Carpenter-Warren, R. Saxena, M. Y. Wong, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, Y. Olivier, E. Zysman-Colman and A. Köhler, J. Phys. Chem. B, 2022, 126, 552–562 CrossRef CAS PubMed.
- X.-Y. Zeng, Y.-Q. Tang, J.-X. Zhou, K. Zhang, H.-Y. Wang, Y.-Y. Zhu, Y.-Q. Li and J.-X. Tang, ACS Appl. Mater. Interfaces, 2024, 16, 16563–16572 CrossRef CAS PubMed.
- C. Montanari, N. Ji Tiwari, R. Misra and B. Carlotti, Chem. – Eur. J., 2024, 30, e202402294 CrossRef CAS PubMed.
- C. Montanari, T. Bianconi, M. Sheokand, T. Teunens, G. Cavalletti, J. Cornil, R. Misra and B. Carlotti, J. Mater. Chem. C, 2023, 11, 10893–10904 RSC.
- H.-Y. Chih, Y.-W. Chen, Y.-C. Hsieh, W.-C. Li, C.-W. Liao, C.-H. Lin, T.-Y. Chiu, W.-W. Tsai, C.-W. Lu and C.-H. Chang, Chem. – Eur. J., 2019, 25, 16699–16711 CrossRef CAS PubMed.
- S. Kumar, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin and E. Zysman-Colman, Asian J. Org. Chem., 2020, 9, 1277–1285 CrossRef CAS.
- T. Serevičius, T. Nakagawa, M.-C. Kuo, S.-H. Cheng, K.-T. Wong, C.-H. Chang, R. C. Kwong, S. Xia and C. Adachi, Phys. Chem. Chem. Phys., 2013, 15, 15850 RSC.
- D. Sun, C. Si, T. Wang and E. Zysman-Colman, Adv. Photonics Res., 2022, 3, 2200203 CrossRef CAS.
- X.-F. Tan, P.-P. Wang, L. Lu, O. Bezvikonnyi, D. Volyniuk, J. V. Grazulevicius and Q.-H. Zhao, Dyes Pigm., 2020, 173, 107793 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- D. Sun, E. Duda, X. Fan, R. Saxena, M. Zhang, S. Bagnich, X. Zhang, A. Köhler and E. Zysman-Colman, Adv. Mater., 2022, 34, 2110344 CrossRef CAS PubMed.
- I. Marghad, F. Bencheikh, C. Wang, S. Manolikakes, A. Rérat, C. Gosmini, D. Hyeon Kim, J.-C. Ribierre and C. Adachi, RSC Adv., 2019, 9, 4336–4343 RSC.
- I. Marghad, D. H. Kim, X. Tian, F. Mathevet, C. Gosmini, J.-C. Ribierre and C. Adachi, ACS Omega, 2018, 3, 2254–2260 CrossRef CAS PubMed.
- S. A. Elgadi, D. M. Mayder, R. Hojo and Z. M. Hudson, Adv. Opt. Mater., 2023, 11, 2202754 CrossRef CAS.
- D. Sun, R. Saxena, X. Fan, S. Athanasopoulos, E. Duda, M. Zhang, S. Bagnich, X. Zhang, E. Zysman-Colman and A. Köhler, Adv. Sci., 2022, 9, 2201470 CrossRef CAS PubMed.
- L. Fisher Jr, R. J. Vázquez, M. Howell, A. K. Muthike, M. E. Orr, H. Jiang, B. Dodgen, D. R. Lee, J. Y. Lee, P. Zimmerman and T. I. Goodson, Chem. Mater., 2022, 34, 2161–2175 CrossRef CAS.
- R. J. Vázquez, H. Kim, P. M. Zimmerman and T. Goodson, J. Mater. Chem. C, 2019, 7, 4210–4221 RSC.
- R. J. Vázquez, J. H. Yun, A. K. Muthike, M. Howell, H. Kim, I. K. Madu, T. Kim, P. Zimmerman, J. Y. Lee and T. G. Iii, J. Am. Chem. Soc., 2020, 142, 8074–8079 CrossRef PubMed.
- T. Bianconi, A. Cesaretti, P. Mancini, N. Montegiove, E. Calzoni, A. Ekbote, R. Misra and B. Carlotti, J. Phys. Chem. B, 2023, 127, 1385–1398 CrossRef CAS PubMed.
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933–9943 CrossRef CAS PubMed.
- D.-G. Chen, T.-C. Lin, Y.-A. Chen, Y.-H. Chen, T.-C. Lin, Y.-T. Chen and P.-T. Chou, J. Phys. Chem. C, 2018, 122, 12215–12221 CrossRef CAS.
- E. Balanikas, T. Bianconi, P. Mancini, N. Ji Tiwari, M. Sheokand, R. Misra, B. Carlotti and E. Vauthey, Chem. Sci., 2025, 16, 8443–8453 RSC.
- M. Söderberg, B. Dereka, A. Marrocchi, B. Carlotti and E. Vauthey, J. Phys. Chem. Lett., 2019, 10, 2944–2948 CrossRef PubMed.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed.
- F. Ricci, B. Carlotti, B. Keller, C. Bonaccorso, C. G. Fortuna, T. Goodson, F. Elisei and A. Spalletti, J. Phys. Chem. C, 2017, 121, 3987–4001 CrossRef CAS.
- P. K. Gupta, F. Khan and R. Misra, J. Org. Chem., 2023, 88, 14308–14322 CrossRef CAS PubMed.
- J. Wang, G. Ouyang, Y. Wang, X. Qiao, W.-S. Li and H. Li, Chem. Commun., 2020, 56, 1601–1604 RSC.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- Y. Wang, G. S. He, P. N. Prasad and T. Goodson, J. Am. Chem. Soc., 2005, 127, 10128–10129 CrossRef CAS PubMed.
- F. Ortica, A. Romani and G. Favaro, J. Phys. Chem. A, 1999, 103, 1335–1341 CrossRef CAS.
- X. Yin, Y. He, X. Wang, Z. Wu, E. Pang, J. Xu and J. Wang, Front. Chem., 2020, 8(725), 1–23 Search PubMed.
- R. Dhali, D. K. A. Phan Huu, F. Terenziani, C. Sissa and A. Painelli, J. Chem. Phys., 2021, 154, 134112 CrossRef CAS PubMed.
- Ó. Guzmán-Méndez, M. M. Reza, B. Meza, J. Jara-Cortés and J. Peón, J. Phys. Chem. B, 2023, 127, 5655–5667 CrossRef PubMed.
- E. F. Plaza-Medina, W. Rodríguez-Córdoba and J. Peon, J. Phys. Chem. A, 2011, 115, 9782–9789 CrossRef CAS PubMed.
- W. Rodríguez-Córdoba, L. Gutiérrez-Arzaluz, F. Cortés-Guzmán and J. Peon, Chem. Commun., 2021, 57, 12218–12235 RSC.
- J. Yang, M. Fang and Z. Li, Aggregate, 2020, 1, 6–18 CrossRef.
- J. Guo, J. Fan, L. Lin, J. Zeng, H. Liu, C.-K. Wang, Z. Zhao and B. Z. Tang, Adv. Sci., 2019, 6, 1801629 CrossRef PubMed.
- X. Chen, S. Bagnich, R. Pollice, B. Li, Y. Zhu, R. Saxena, Y. Yin, W. Zhu, A. Aspuru-Guzik, E. Zysman-Colman, A. Köhler and Y. Wang, Adv. Opt. Mater., 2024, 12, 2301784 CrossRef CAS.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., 2018, 130, 16645–16649 CrossRef.
- W. Dai, T. Bianconi, E. Ferraguzzi, X. Wu, Y. Lei, J. Shi, B. Tong, B. Carlotti, Z. Cai and Y. Dong, ACS Mater. Lett., 2021, 1767–1777 CrossRef CAS.
Footnote |
| † Equally contributed as first authors. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Benedetta
Carlotti
*b and
Benedetta
Carlotti










