Spacer-directed comparable and contrasting optical properties in D–A molecules enabling the fabrication of efficient novel thin-film phototransistors
Received
30th July 2025
, Accepted 3rd November 2025
First published on 4th November 2025
Abstract
Intuitive and insightful reasoning based on experimental evidence can be employed for devising a hypothesis aimed at developing functional molecules equipped with the desired optical properties. Using this rationale, the present study details a bimodal strategy for creating contrasting and comparable optical properties using novel donor–acceptor type molecules. The study relied on the distinct architecture of curcumin-BF2-based D–A molecules, which was achieved by varying the spacers from rigid (conjugated; D–π–A) to flexible (conjugation forbidden; D–σ–A) as well as varying the strength of the donor species from anthracene to carbazole. Four molecules, AnBr/CbBr (D–π–A) and AnBf/CbBf (D–σ–A), were designed and synthesized. The combined synthetic, steady-state and time-resolved photophysical and theoretical approaches established the contrasting emissive behaviors of the D–π–A and D–σ–A systems, indicating that the former was more emissive in solution (aggregation-caused quenching) and the later was more emissive in the solid state (aggregation-induced emission). The comparative optical behaviors observed were achieved by varying the strength of the donor, which led to strong dual emission in AnBf that contrasted with that of CbBf. However, the higher-emission quantum yield of CbBr relative to AnBr demonstrated the strong electron-donating ability of the carbazole. Owing to their specific absorption in the solid state, AnBf and CbBf were further employed for the fabrication of blue-sensitive photodetectors. These photodetectors were fabricated in a phototransistor geometry, which showed a large variation in the threshold voltage and off-current of the devices. The devices with the CbBf and AnBf molecules showed excellent photoresponsivities of 3 A W−1 and 1.4 A W−1, respectively, for blue light with an intensity of 15 W m−2.
Introduction
In recent years, extensive studies on the excited-state properties of organic, donor–acceptor (D–A) fluorophores have shown that the design of novel organic materials for applications in photovoltaics,1–3 photodynamic therapy (PDT),4–10 luminescence bioimaging,11,12 and artificial photosynthesis13–15 depends on the way we process and modulate charge transfer (CT) events. Non-radiative charge separation and radiative charge recombination have been the focus of such modulation of CT events and serve as effective tools that influence the design principles of D–A fluorophores.16–20 This class of materials is broadly classified into three categories; (i) single head donor–acceptor chromophores (D–A), (ii) double head donor–acceptor–donor (D–A–D) chromophores, and (iii) double-head acceptor–donor–acceptor (A–D–A) chromophores.
Under illumination, intramolecular charge transfer (ICT) can occur, allowing for the separation of photogenerated charge carriers (electrons and holes) within molecules, thereby providing additional control over their optoelectronics properties.21,22 The observation that intramolecular CT is a dominant transition that plays a crucial role in governing photophysical properties and efficient CT has been evidenced over short- and long-range distances in D–A fluorophores.23,24 This is most apparent in the cases where the strong donor (D) and acceptor (A) moieties are connected by a π-linker in a single molecular framework. However, weak donor/acceptor or multiple donor and acceptor moieties are now being appended for deriving ancillary information about CT processes.25–29 Organic phototransistors have emerged as a promising class of light-sensing devices due to their mechanical flexibility, low-cost fabrication, and tunable optoelectronic properties. Among the various materials explored, donor–acceptor-(D–A)-based conjugated molecules are particularly attractive, offering high photoresponsivity and spectral tunability due to their rational molecular design.30–33 In D–A systems, an electron-donating unit (D) is covalently linked to an electron-accepting unit (A) via a π-conjugated bridge, enabling intramolecular charge transfer (ICT) upon light absorption. This architecture facilitates broadband absorption, reduced bandgaps, and enhanced charge separation, all of which are crucial for efficient phototransistor operation. By strategically engineering the donor and acceptor components, key properties, such as the HOMO–LUMO energy levels, optical absorption range, and carrier mobility, can be finely tuned to meet application-specific requirements. In phototransistors, these tailored D–A materials function as the active semiconductor layer that converts incident photons into an amplified electrical signal. Their unique ability to combine light sensitivity with intrinsic gains makes them ideal for applications, such as optical sensing, artificial vision systems, and low-light detection. Moreover, the versatility of D–A chemistry allows for the integration of D–A materials into both small molecule and polymer systems, broadening their applicability across different device architectures, including top-gate and bottom-gate field-effect transistors.34,35 Recent advances in D–A molecular design have led to the development of high-performance phototransistors with responsivities exceeding 103 A W−1, detectivities in the 1012 Jones range, and fast response times under visible and near-infrared (NIR) illumination. These improvements stem from innovations in molecular planarity, side-chain engineering, and the incorporation of strong acceptors, such as benzothiadiazole, diketopyrrolopyrrole (DPP), and perylenediimide (PDI).36,37
This study aims to explore the fundamental photophysical characteristics of D–A conjugated molecules and their relationship to phototransistor performance. We also intend to highlight recent progress in material design, strategies for device fabrication, and the application-specific implementations of D–A-based phototransistors in the broader field of organic electronics. The emerging concept of aggregation-induced emission (AIE), which is based on altering the conventional aggregation-caused quenching (ACQ) luminophores into AIE luminogens has led to improvements in the fluorescence performance of D–A luminophores and the realization of their multifarious applications.38–40 AIE largely depends on the rigidification of flexible molecular structures in the excited state, which restrains the non-radiative decay pathways and enhances fluorescence. A comprehensive regulatory framework has already been established to study electronic effects on AIE and it has demonstrated that increasing the strength of either the D or A units facilitates color tunability in AIEgens.41–43 Furthermore, it has recently been demonstrated that space-enough and conjugation-forbidden linkages between the D and A units can lead to intermolecular transitions, which can further improve emissive properties when coupled with the inherent intramolecular transitions in D–A based AIEgens.44–46 Thus, it can be surmised from these aforementioned facts that the design of an AIEgen requires appropriate preorganization of the donor and acceptor moieties, and that the relative positioning of the D and A units, along with their respective strength, affects the outcomes of electronic coupling. In this context, a molecular system can be designed and constructed that provides a platform where optical behavior can be compared and contrasted, which can further lead to a more precise development of molecules for various applications. To streamline the process and manage the complexity of molecular designs and extensive preparation procedures, we chose a β-diketone difluoride core for its ease of preparation and flexibility in synthetic manipulations.
A bimodal strategy was devised, allowing for the comparison and contrast of optical behaviour. Firstly, the strategy relied on varying the type of spacer, which allowed for a comparison between the emission in solution and in the solid state. Secondly, the strategy allowed for a change in the type of donor, which allowed us to attain differential optical properties. In this direction, the β-diketone difluoride platform has been used as acceptor to construct D–A type systems with conjugated and conjugation-forbidden linkages; simultaneously, the types of donors were also changed from anthracene to carbazole in order to assess the resultant electronic coupling. In this series, the D and A units were linked with a conjugated bridge to give AnBr and CbBr (D–π–A), while the non-conjugated linkage between the D and A units yielded compounds AnBf and CbBf (D–σ–A). These linkages provided a rigidified core in AnBr and CbBr, while the methylene units conferred the desired flexibility to AnBf and CbBf. The flexible systems were found to display AIE characteristic along with significant solid-state emission, while the rigid molecules displayed strong emission in a dilute solution, the ACQ effect and diminished solid-state emission. On the other hand, the cumulative effect of the spacer and donors contributed to comparable charge-transfer properties, which is an inherent trait of D–A molecules. The limited exploration of AIEgens as interfacial materials for electrodes47 prompted us to evaluate the applicability of AnBf and CbBf in organic electronic devices by fabricating blue-sensitive photodetectors. The photodetectors were fabricated based on a phototransistor geometry, where the channel of the transistor was made of an organic/inorganic bilayer structure. The device fabrication and photoresponsivity measurements are detailed herein.
Experimental methods
Reagents
9-Anthraldehyde, 9H-carbazole, 4-hydroxybenzaldehyde and boron trifluoride etherate were purchased from Sigma-Aldrich, India. Common reagents, potassium hydroxide, sodium hydroxide, trifluoroacetic acid, triethylamine and the solvents, ethyl acetate, hexane, dimethylsulphoxide (DMSO), dichloromethane (DCM), tetrahydrofuran (THF), dimethylformamide (DMF) and methanol were procured from Avra Chemicals Hyderabad, India; they were dried and distilled following standard procedures reported in the literature.48 The synthetic manipulations were performed under an oxygen-free, nitrogen atmosphere and the photophysical studies were done using spectroscopic-grade solvents.
General information
1H (500 MHz), and 13C (125 MHz) NMR spectra were acquired on JEOL AL 500 FT-NMR spectrometer at room temperature using Si(CH3)4 as an internal reference. The UV-vis and emission spectra were acquired at room temperature on a Shimadzu UV-1800 and PerkinElmer LS55 Fluorescence spectrometers, respectively. Electrospray ionization mass spectrometric (ESI-MS) measurements were acquired on a Bruker-Daltonics mass spectrometer. Time-resolved fluorescence lifetime experiments were performed on a TCSPC system from Horiba Yovin (Delta Flex). The compounds were excited at 482 nm using a pico-second diode laser (Model: Delta Diode). Data analyses were then performed using the decay analysis software (HORIBA Scientific: EzTime). The crystallographic data based on X-ray intensity were acquired for the AnBr crystals with well-defined shapes using different detectors. Specifically, the XtaLAB synergy dual flexhypix 3000 CCD detector was used for AnBr. The X-ray radiation source utilized for all the crystals was Mo–Kα (l = 0.71073 Å). The crystal structures were determined and refined through a process involving direct methods (SHELXS 97) and subsequent full-matrix least-squares refinement on F2 (SHELX 14) within the OLEX2 environment.49–52 The anisotropic assignment of all the atoms, excluding hydrogen (H), was carried out. Analyses of the interaction and stacking distances were performed using PLATON.53 The crystallographic data for this study in the CIF format can be found under the CCDC deposition number 2339002 (AnBr).
Procedure to develop PMMA film
The PMMA film was developed following a procedure reported in the literature.46 In short, a solution of PMMA (200 mg) in 2 mL THF was heated at 60 °C with continuous stirring. After heating, 20 µL of the compounds (c, 50 µM) in THF was added to the resulting viscous solution of PMMA, while stirring was maintained at the same temperature for 2 h. The mixture was then transferred into glass molds and left to slowly dry at room temperature.
Device fabrication
To demonstrate an applicability of the solid-state optical absorption imparted by the unique D–σ–A construction of AnBf and CbBf, photodetectors were fabricated. These photodetectors were fabricated with a phototransistor geometry, where the channel of the transistor was made of an organic/inorganic bilayer structure, while the bottom tin oxide layer acted as the primary charge-transport layer and the organic AnBf or CbBf molecules acted as the blue-light absorbing layer. When light was illuminated on the channel, the organic molecules primarily absorbed the photons and subsequently produced photogenerated electrons and holes. Due to the relatively wider bandgap of the inorganic SnO2, the bottom layer was unable to generate much photocurrent as was observed from the SnO2-only reference phototransistor device. Out of the additional carriers, the photogenerated electrons were transferred to the SnO2 layer, while the holes remained in the organic layer. Overall, due to these additional carriers, the channel conductivity increased largely with light intensity. The variation of the channel conduction allowed us to detect the light intensity. The experimental details pertaining to material synthesis, device fabrication and electrochemical characterization have been provided in the SI. The reference SnO2 thin-film transistors (TFT) was named device-1, while the TFTs coated with CbBf and AnBf were named device-2 and device-3, respectively (Fig. S1).
Syntheses
The syntheses of A1, 9-methyl-9H-carbazole-3-carbaldehyde (2), 4-(2-(9H-carbazol-9-yl)ethoxy)benzaldehyde (Cb1) 4-(anthracen-9-ylmethoxy)benzaldehyde (An1) were achieved according to previously reported procedures.54–56
Synthesis of A1.
In a 100-mL round bottom flask, 10 mL DCM was mixed with 4 mL acetyl acetone (38.94 mmol) and then 5 mL BF3OEt2 (38.94 mmol) was added dropwise for 30 min and the reaction mixture was refluxed overnight. At the end of the reaction, the reaction solution was extracted with DCM/H2O (50 mL × 3). The organic layer was separated, dried over anhydrous Na2SO4 and evaporated under vacuum. The crude product obtained was then purified by column chromatography (SiO2; ethyl acetate/hexane), yielding compound A1 as a brown solid (2.8 g, 70.0% yield). 1HNMR (CDCl3, 500 MHz, δ ppm): 5.998 (s, 1H), 2.4 (s, 6H). 13CNMR (CDCl3, 125 MHz, δ ppm): 101.9, 77.25, 77.00, 76.74, 23.99. 11B NMR (160.4 MHz, CDCl3, δ ppm): −0.316. 19F NMR (470.6 MHz, CDCl3, δ ppm): −138.034, −138.097.
Synthesis of CbBr.
To a two-necked round bottom flask, 2.2 mmol of aldehyde 2 and 1.0 mmol of A1 were dissolved in a minimum amount of ethyl acetate, and the flask was flushed with nitrogen. N-Butylamine (0.22 mmol) was then added dropwise to the reaction mixture via a syringe, and the reaction mixture was stirred overnight at room temperature. After the reaction was completed, a black-colored solid precipitated, which was filtered through a Buchner funnel, washed with cold ethyl acetate and diethyl ether, and dried under vacuum. This process yielded CbBr as a black-colored solid (yield: 76.2%). 1H NMR (500 MHz, CDCl3, δ ppm): 8.84 (s, 1H), 8.14 (d, J = 7.4 Hz, 3H), 7.57 (t, J = 7.7 Hz, 1H), 7.43 (dd, J = 18.7, 8.6 Hz, 3H), 7.36 (t, J = 7.4 Hz, 1H), 6.63 (s, 1H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3, δ ppm): 188.8, 182.8, 145.4, 141.9, 127.4, 127.2, 123.3, 122.9, 121.5, 121.2, 121.0, 109.5, 109.0, 96.6, 29.6. C33H25BF2N2O22+ [M + H]+ 531.19; found 531.19.
Synthesis of CbBf.
This compound was synthesized following the procedure for CbBr using A1 and Cb1. CbBf was obtained as a bright-orange powder (yield: 67.1%).1H NMR (500 MHz, DMSO-d6, δ ppm): 8.14 (d, J = 6.0 Hz, 4H), 7.91 (d, J = 15.8 Hz, 2H), 7.72 (dd, J = 27.8, 6.7 Hz, 8H), 7.47 (s, 5H), 7.21 (s, 4H), 7.01–6.91 (m, 4H), 6.51–6.47 (m, 2H), 4.83 (s, 4H), 4.47 (s, 4H). 13C NMR (126 MHz, DMSO-d6, δ ppm): 67.32, 110.12, 115.36, 119.42, 119.48, 120.68, 122.70, 126.20, 127.45, 127.45, 127.45, 127.60, 132.17, 132.28, 140.77, 146.83, 161.88, 179.75. C47H37BF2N2O42+ [M + H]+ 743.28; found 743.27.
Synthesis of AnBr.
This compound was synthesized following the procedure for CbBr using A1 and 9-anthraldehyde. AnBr was obtained as a dark blackish powder (yield: 70.4%). 1H NMR (500 MHz, DMSO d6, δ ppm): 9.13 (s, 1H), 9.10 (s, 1H), 8.80 (s, 1H), 8.37 (d, J = 8.7 Hz, 4H), 8.20 (d, J = 8.3 Hz, 4H), 7.71–7.67 (m, 4H), 7.64–7.61 (m, 4H), 7.25 (s, 1H), 7.16 (s, 1H), 7.13 (s, 1H), 6.51 (s, 1H). 13C NMR (126 MHz, DMSO-d6δ ppm): 102.86, 106.58, 125.36, 126.40, 128.13, 128.85, 129.47, 129.91, 130.58, 130.81, 131.42, 135.05, 144.11, 180.59. C35H23BF2O22+ [M + H]+ 525.17; found 525.18.
Synthesis of AnBf.
This compound was synthesized following the procedure for CbBr using A1 and An1. AnBf was obtained as a bright-orange powder (yield: 65.3%).1H NMR (500 MHz, DMSO d6, δ ppm): 5.56 (2H, s), 6.07 (1H, s), 7.36 (4H, d, J = 6.5 Hz), 7.47 (4H, d, J = 6.5 Hz), 7.55–7.7 (10H, m), 8.23 (4H, t, J = 8.0 Hz), 8.81 (4H, d, J = 7.0 Hz), 9.49 (4H, d, J = 5.5 Hz). 13C NMR (126 MHz, DMSO d6, δ ppm): 63.20, 101.59, 116.26, 118.56, 124.63, 125.85, 127.34, 129.42, 129.52, 130.99, 131.68, 132.39, 148.50, 163.03, 166.69, 181.66. C49H35BF2O42+ [M + H]+ 737.61; found 737.54.
Results and discussion
We designed and synthesized two series of D–A constructs: the first series featured two D–A molecules (AnBr, CbBr) with a fully conjugated, rigid scaffold, while the second series comprised two D–A systems (AnBf, CbBf) with a forbidden conjugation and flexible scaffold. The donor strength was assessed by varying the donors between anthracene (AnBr, AnBf) and the carbazoles (CbBr, CbBf); in both series, the A unit was kept constant. As shown in Schemes 1 and 2, all the compounds were prepared in moderate-to-good yields through multistep synthetic pathways. The rigid systems, AnBr and CbBr, were prepared through the Knoevenagel condensation of 9-anthraldehyde and 9-methyl-3-carbazolecarbo-xaldehyde with A1, respectively. For AnBf and CbBf, a more detailed and rather intricate synthetic plan was followed. Firstly, the aldehydes An1 and Cb1 were synthesized following methods in the literature54–56 and were further condensed with A1, affording AnBf and CbBf. All the compounds were characterized using 1H NMR, 13C, 11B, 19F NMR spectroscopic techniques and mass spectrometry (Fig. S3–S12).
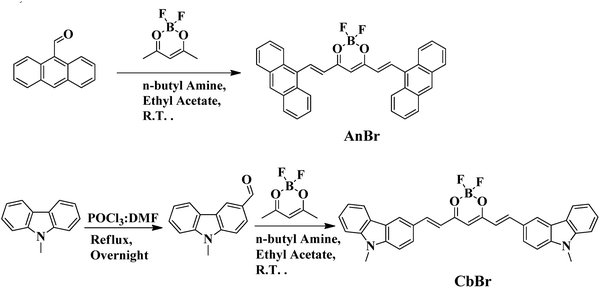 |
| | Scheme 1 Synthetic route of the rigid systems, AnBr and CbBr. | |
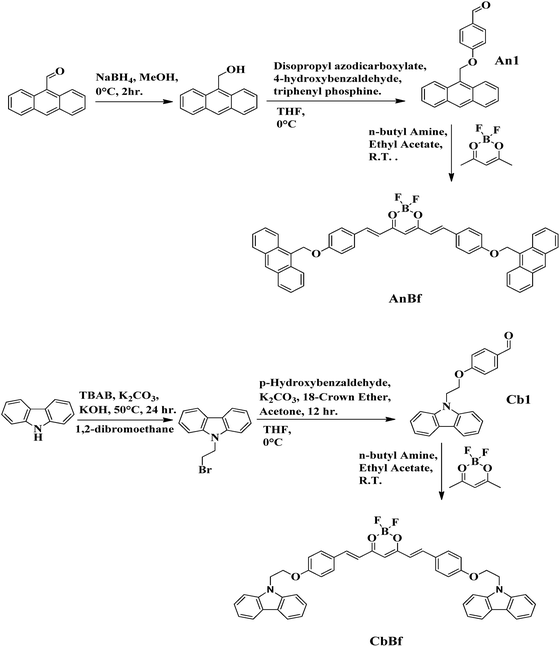 |
| | Scheme 2 Synthetic route of the flexible systems, AnBf and CbBf. | |
The absorption and photoluminescence properties of all the compounds were thoroughly investigated in solution and in the solid and aggregated states. To gain a deep insight into the occurrence of the intermolecular and intramolecular charge-transfer states, the optical profiles of anthracene and N-methyl carbazole were determined and compared with that of their synthesized analogues (Fig. S16). The UV-vis absorption spectrum of AnBf displays an absorption maximum at 395 nm, while that of AnBr exhibits a broad absorption band at 534 nm along with a less intense band at 366 nm. When compared with the absorption spectrum of 9-anthraldehyde, the band at 395 nm in the absorption spectrum of AnBf corresponds to an absorption maximum associated with the donor unit at 400 nm (Fig. 1(a)). CbBf displays an absorption maximum at 484 nm along with a comparatively less intense band at 459 nm; however, CbBr exhibits a significantly red-shifted absorption band at 537 nm along with a shoulder at 505 nm. Both CbBf and CbBr display weak absorption bands at ∼340 nm, which correspond to the absorption band of the carbazole unit (Fig. S16). These results indicate that in presence of the weaker donor, i.e. anthracene, there were no intramolecular interactions between the D and A units in AnBf, while these same interactions were responsible for the red-shifted absorption band in AnBr. On the other hand, despite the non-conjugated bridge between the D and A units in CbBf, there was some charge-transfer interaction, which produced a red-shifted absorption band; however, the stronger D–A interactions present in CbBr relative to CbBf due to the conjugated bridge caused an increased red-shifted absorption in CbBr. A close inspection of the emission spectra reveals a rather complex emission behavior of the synthesized D–A molecules. Upon excitation at 395 nm, AnBf displays dual emission bands at 469 and 515 nm. On the other hand, CbBf displays a very weak emission, peaking at 519 nm. Furthermore, the emission profiles of AnBr and CbBr display emission maxima at 635 and 588 nm after excitation at 534 and 537 nm respectively. These results clearly evidence that the emission intensity of AnBr was much less than that of CbBr (Fig. 1(b)).
 |
| | Fig. 1 UV-vis absorption (a) and emission (b) spectra of CbBr, CbBf, AnBr and AnBf in THF (c = 30 µM). | |
The intended contrast in the optical behavior of the D–σ–A systems was evidenced in their blue-shifted emission compared to their respective D–π–A systems; this difference was attributable to the increased π-conjugation in the D–π–A systems. Furthermore, a comparative difference in the optical profiles was observed in the emission profiles of AnBr/CbBr and AnBf/CbBf. CbBr exhibited a more intense emission relative to AnBr owing to its greater electron-donating capability of the carbazole donors. However, contrary to our expectation, AnBf displayed a more intense emission and dual emission bands when compared with CbBf. This deviation from our expectation likely occurred not only because of the flexible construction of AnBf but due to excimer formation and an intermolecular CT, which accounted for the dual emission bands. To verify the formation of an excimer as well as differentiate between the CT and excimer emission bands, we further recorded the emission spectra of AnBf at various concentrations in THF (Fig. 2(a)). The spectra revealed that in very dilute solutions of AnBf (c = 10−7 M), the band at 469 nm was more intense than the band at 515 nm; however, as the concentration of AnBf in THF increased, the band at 469 nm started to decreased, while the intensity of the band at 515 nm increased. Furthermore, when the concentration of AnBf increased to 10−4 M, the 515-nm band became dominant, while the intensity of the higher-energy band was significantly reduced. This observation suggests that the excimer was formed at higher concentrations, and thus the lower-energy band at 515 nm was attributed to the excimer and the higher-energy band at 469 nm corresponded to a CT state in AnBf.57
 |
| | Fig. 2 Emission spectra of AnBf at different concentrations in THF (a) and emission spectra of AnBf in the PMMA film at varying concentrations of PMMA from 1 to 20 mg mL−1 (b). | |
Solvatochromism
The D–A construction of the molecules in this study inherently endowed them with charge-transfer properties and the formation of the CT state was verified by examining their absorption (Fig. S13) and emission spectra (Fig. 3) in a diverse array of solvents with distinct polarities (benzene, toluene, 1,4-dioxane, CHCl3, THF, and DCM). It was observed that the ground state remains impervious under varying solvent polarities as the absorption spectra demonstrated insignificant changes. Contrastingly, the emission spectra showcased significant variation along a solvent polarity gradient. As expected, the D–π–A systems comprising CbBr and AnBr displayed a pronounced positive solvatochromism with bathochromic shifts of 46 nm and 35 nm, respectively, along a gradient characterized by a transition from non-polar to polar solvents. On the other hand, CbBf and AnBf exhibited substantial bathochromic shifts of 39 nm and 44 nm, respectively, under a similar solvent gradient. The solvatochromic shift of CbBf was less than that of CbBr, which is consistent with the reduced conjugation in the latter system (Fig. 3 and Table S2). Noteworthy is the solvatochromic behavior of AnBf, which displays a higher solvatochromic shift relative to its rigid counterpart, AnBr. The formation of the intermolecular CT state, which is a common occurrence in conjugation-forbidden D–A molecules, can account for this anomaly.49 To further deepen our understanding of the intermolecular CT state in AnBf, we further acquired the emission spectra of AnBf (c = 50 µM) under varying concentrations of PMMA from 1 to 20 mg mL−1. Apparently, the intensity ratios for the emission maxima at 469 and 515 nm were found to be 1.057, 0.883, 0.816, 0.704 and 0.778 at PMMA concentration of 1, 5, 10, 15 and 20 mg mL−1, respectively. The emission maxima at 20 mg mL−1 of PMMA was very similar to the maxima obtained at a high concentration (c = 100 µM), supporting the formation of an excimer and an intermolecular CT state (Fig. 2(b)).57
 |
| | Fig. 3 Normalized emission spectra of CbBr (a), CbBf (b), AnBr (c) and AnBf (d) (c = 30 µM) in solvents of varying polarity. | |
For a comprehensive assessment of the ICT in CbBr, AnBr, CbBf, and AnBf, the fluorescence quantum yield of each compound was systematically calculated across varying solvent polarities (Table S2). As expected, there was a notable increase in the quantum yield in toluene (CbBr: 26.7%, AnBr: 30.5%, CbBf: 46.2%, and AnBf: 30.5%) compared to DCM (CbBr: 10.7%, AnBr: 5.1%, CbBf: 0.84%, and AnBf: 4.4%). This substantial decrease in polar solvents strongly suggests a shift towards non-radiative relaxation processes.
Aggregation-induced emission
In order to further validate the desired outcome of the bimodal strategy, we investigated the absorption and emission of AnBr/CbBr and AnBf/CbBf in the aggregated and solid states (Fig. S14, S15, S20 and S21). The aggregated-state emission behavior was monitored by recording the emission profiles of AnBr/CbBr and AnBf in THF/water (c = 30 µM) with varying water fractions (fw) from 0 to 99.9%, while that of CbBf was done in THF/hexane (c = 30 µM) with varying hexane fractions (fH) from 0 to 99.9% (Fig. 4). As expected, the rigid systems, AnBr and CbBr with fully conjugated scaffolds suffered from severe aggregation-caused quenching (ACQ) and displayed insignificant emission in the aggregated state. On the other hand, the close observation of the emission behavior of AnBf with increasing fw revealed an initial emission quenching up to fw = 70%; however, the dual emission bands were found to be intact at this point. With a further increase in fw to 80%, the band at 469 nm decreased in intensity, while the one at 515 nm shifted to 560 nm; however, the emission intensities were still relatively lower than the original intensity at fw = 0%. Upon increasing the fw to 99.9%, the higher-energy band at 515 nm almost disappeared and the red-shifted band at 560 nm displayed a significant emission enhancement (∼2.5 times). The disappearance of the band at 515 nm can be attributed to the loss of excimer emission in the aggregated state, while the red shift in the CT band can be related to increased intermolecular interactions in the aggregated state. Further, due to the extremely poor solubility of CbBf in water, aggregate formation was achieved in THF/hexane. CbBf displayed an ∼8 times emission enhancement of the band at 519 nm at fH = 80%. A further increase in fH led to emission quenching due to deceased solubility at a higher fH. These observations categorize AnBf and CbBf as efficient AIEgens. It is quite fascinating that the emission enhancement of CbBf was much greater than that of AnBf, which can be attributed to the strong electron-donating capability of carbazole. In addition, the AIE behavior was further authenticated through fluorescence lifetime measurements (Fig. S17). As a result of the AIE effect, an increase in the fluorescence lifetime from 0.22 ns (fw = 0%) to 0.43 ns (fw = 99%) was observed for AnBf, while CbBf displayed an increase in lifetime from 0.18 ns (fH = 0%) to 0.38 ns (fH = 80%). Further, the SEM images revealed the formation of spherical aggregates for AnBf and CbBf (Fig. S18).
 |
| | Fig. 4 Emission spectra of CbBf (a) and AnBf (b) in THF, THF/n-hexane and THF/water as a mixture (c = 30 µM) with increasing fractions of n-hexane and water. | |
In accordance with the emission in the aggregated state, we further examined the solid-state emission behavior and tried to verify whether the bimodal strategy stood up to solid-state rigidification, owing to its increased intermolecular interactions. The results were quite interesting as AnBr and CbBr were found to be non-emissive in the solid state (Fig. 5). On the other hand, AnBf and CbBf displayed significant emissions with emission maxima centered at 648 and 684 nm with photoluminescence quantum yields (PL-QY) of 56.19% and 13.28%, respectively. Notably, the stronger donor, i.e. carbazole, led to a more red-shifted emission in the solid state for CbBf, reciprocating the comparative optical behavior.
 |
| | Fig. 5 Solid-state emission spectra of CbBf (a) and AnBf (b) (λex = 380 and 370 nm, respectively); the insets show the solid-state emission of CbBf and AnBf under UV light (λex = 365 nm). | |
We were successful in obtaining the single crystals of AnBr by the slow evaporation of the CH2Cl2/hexane solution over a period of three weeks. The single-crystal structure of AnBr was verified by X-ray single-crystal studies and OLEX2 views, as shown in Fig. S19. It crystallizes in the monoclinic crystal system with the P121/n1 space group. The crystal packing and data refinement parameters for AnBr have been provided in Table S3. It has been observed that AnBr exists in the form of a dimer through a single π–π interaction with a distance of 3.379 Å.
Density functional theory calculations
Theoretical investigation through the geometrical optimization of the (D–A) systems were performed using the B3LYP method with the 6-31G** basis set. Fig. 6 illustrates the distribution of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) for CbBr, CbBf, AnBr and AnBf. As can be seen in Fig. 6, the HOMOs for AnBr and CbBr are centered mostly over the donor anthracene and carbazole units, respectively, while the LUMO is largely located on the acceptor difluoroboron moiety. It is worth mentioning that the conjugated π-bridge participates in both the HOMO and LUMO distributions. On the other hand, the HOMOs in AnBf and CbBf are located exclusively on the anthracene and carbazole moieties, while the LUMO is spread over the difluoroboron moiety. In the D–σ–A systems, the alkyl spacer does not participate either in the HOMO or the LUMO distribution. The complete breakage of the conjugation between the D and A units in AnBf/CbBf suggests that the suppression of ICT contrasts with what is commonly observed in most D–A systems. Furthermore, a reduced HOMO–LUMO overlap was found in AnBf and CbBf relative to AnBr and CbBr, which further suggests a suppression of the ICT in flexible systems (D–σ–A) as compared to rigid (D–π–A) compounds. Furthermore, the UV-vis spectra of CbBr, CbBf, AnBr and AnBf were obtained from the TD-DFT calculations (Fig. S19 and Table S1) and these data show good agreement with our experimental data. Table S1 summarizes the details of the absorption wavelength, energy, oscillation strength (f), assignment, and transitions.
 |
| | Fig. 6 HOMO–LUMO frontier molecular orbital (FMO) diagram of CbBr, CbBf, AnBr and AnBf. | |
Electrical and optical response of TFTs
The output (ID–VD) (drain current–drain voltage) and transfer (ID–VG) (drain current–gate voltage) characteristics of device 1 are shown in Fig. S2a and b, respectively, which show n-channel transport of the device. For the output characteristics, the VD was varied from 0 V to 2 V under different constant gate biases within the range from 0.5 V to 2 V. The output characteristics of device 1 showed a very good drain–current saturation within a 2-V drain voltage when a 2-V gate bias was applied, implying that a 2-V external bias was sufficient to drive this TFT. It can be noted that the operating voltage of this TFT was only limited to within 2.0 V, which is due to the high-areal capacitance of such ionic-gate, dielectric thin films.58 During the study of the transfer characteristics, VG was varied from −2 V to 2 V at a fixed VD of 2 V (Fig. S2b). This characteristic shows that the device 1 had a threshold voltage (VTh) of ∼0.5 V with an on/off ratio of 6.3 × 104, which is reasonably good at a 2-V operating voltage for TFTs. The effective carrier mobility (µ) in the saturation region and subthreshold swing (SS) of these TFTs were calculated from the following equations, respectively:59| | 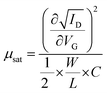 | (1) |
| | 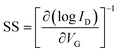 | (2) |
where C, ID, Vth and VG, are the capacitance per unit area, saturation drain current, threshold voltage and gate voltage, respectively. To estimate the carrier mobility using eqn (1), the transfer characteristics were replotted with the x-axis of I1/2D and the mobility was calculated from the slope of that plot, whereas the extrapolation of the slope gave the threshold voltages of the devices (Fig. S2b). The TFT parameters of device 1 are summarized in Table S4. The extracted saturation mobility of device 1 was 1.0 cm2 V−1 s−1, whereas the SS value was 236 mV dec−1. Additionally, the device was illuminated with blue light with different intensities from 0 to 15 W m−2, and the results did not show any significant difference in transfer characteristics, indicating that the reference SnO2 transistor did not show any blue-light sensitivity (Fig. S2d). Similarly, device-1 was also tested under different UV light is illuminations resulting in a significant variation of the drain current as shown in Fig. S2c. The optical behavior of the reference SnO2 TFT implies that the device displays photosensitivity only for high-energy photons (like UV light).
Similarly, electrical and optoelectrical characterizations were done for device-2 and device-3, which are shown in Fig. 7 and 8, respectively. Similar to device-1, both of these bilayer devices showed good current saturation in their output characteristics under a 2.0-V operating voltage (Fig. 7(a) and (a)). Additionally, the on-current of the devices significantly increased compared to the reference devices (device 1). As a consequence, the effective carrier mobilities also increased, yielding 1.5 and 1.1 cm2 V−1 s−1 for device-2 and device-3, respectively. The transfer characteristics of the bilayer devices show that the VTh of the devices shifted towards negative voltages of −0.8 V and 0.1 V for device-2 and device-3, respectively (Fig. 7(b) and (b)). The on/off ratio (under dark) of the TFTs remained almost similar. The optoelectrical response of device-2 shows that the device had a very high photosensitivity both under UV and blue lights, particularly under a depletion-mode operation, as shown in Fig. 7(c) and (d), respectively. Fig. 7(c) shows that the VTh of this device shifted from −0.8 V to −1.4 V when the UV-light intensity increased from 0 to 3 W m−2; this variation was almost linear with the UV-light intensity, as shown in Fig. 7(e). Similarly, the VTh shifted from −0.8 V to −1.15 V when the blue-light intensity increased from 0 to 15 W m−2, displaying linearity in the higher-intensity range. Similar blue-light photosensitivity was also observed in device-3, as shown in Fig. 8(c).
 |
| | Fig. 7 (a) Output and (b) transfer characteristics of device-2 (TFT using the SnO2/CbBf bilayer channel) under dark conditions, (c) under different UV light intensities and (d) under different blue-light intensities; light intensity vs. VTh shifting of device-2 (e) under UV, (f) under blue light. (g) energy band diagram of the SnO2/CbBf heterojunction and associated photogenerated charge transfer. | |
 |
| | Fig. 8 (a) Output and (b) transfer characteristics of device-3 (TFT using the SnO2/AnBf bilayer channel) under dark conditions, (c) under different blue-light intensities, (d) light intensity vs. VTh shifting of device-3, (e) energy band diagram of the SnO2/AnBf heterojunction and associated photogenerated charge transfer. | |
The high photoresponse of these SnO2/organic bilayer channel phototransistors originated from the type-II heterojunction formation at the organic/inorganic interface. The conduction band (CB) and the valence band of this sol–gel-derived SnO2 were 4.0 and 7.5 eV respectively, with a bandgap of 3.5 eV,60 while the HOMO and LUMO levels of the CbBf molecules were 2.63 and 5.52 eV, respectively (Fig. 7(g)). On the other hand, the AnBf molecules had similar HOMO and LUMO levels (Fig. 8(e)). When light was illuminated from the top of the channel, most of the light was absorbed by the organic layer due to its lower bandgap, producing photogenerated electron–hole pairs. The built-in potential of the SnO2/organic interface helped to dissociate the electron–hole pairs and transfer electrons to the SnO2 layer, increasing the electron density of the SnO2 layer, effectively elevating the DC conductivity of the channel and raising the depletion-mode current of the transistor. As the light intensity was increased again, this reverse-biased current increased further, as shown in Fig. 7(c) and (d) for device-2 and in Fig. 8(c) for device-3.
A light-dependent resistor (LDR) is a passive electronic component whose resistance decreases (and current increases) with increasing light intensity. Fig. S23 shows that when the light intensity increased, the current of the device increased. For device-2, at a fixed gate voltage, FVG = −0.75 V, the change in current increased linearly with UV- and blue-light intensities (Fig. S23a and b). Fig. S23c shows that there was a linear change in current as the blue-light intensity increased for device-3.
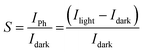
The photosensitivity (S), photoresponsivity (R) and detectivity (D*) of device-2 and device-3 were calculated from the following equations:
| |  | (3) |
| |  | (4) |
| | 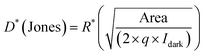 | (5) |
where,
IPh,
Popt, and
Idark are the photocurrent, power of incident light, and the dark current of the device, respectively. Meanwhile,
q,
Ilight, and
h are the electronic charge (1.6 × 10
−19 coulomb), device current under light conditions, and Planck's constant (6.62 × 10
−34 m
2 kg s
−1). The photosensitivity (
S), photoresponsivity (
R) and detectivity (
D*) of device-2 and device-3 under blue-light illumination are presented in
Fig. 9. These data indicate that the photosensitivity (
S) of these devices was very high in a narrow zone of the gate bias, particularly under the flatband conditions (when the accumulated charge in the channel was nearly zero). These photosensitivity data also indicate that the blue-light sensitivity of device-3 was ∼2500 under a 15-W m
−2 illumination (
Fig. 9(d)), which is very high; this sensitivity was more than an order higher than that of device-2 (
Fig. 9(a)). It also can be noted that the photoresponsivity (
R) of these devices are very high under accumulation-mode operation (
Fig. 9(b) and (e)). Moreover, the magnitude of R decreased with light intensity. This responsivity of device-2, which was ∼3 A W
−1, is quite high, more than double that of device-3. Additionally, the detectivity of device-2 and device-3 under accumulation-mode operation was on the order of 10
9 Jones, which is quite high for a phototransistor device (
Fig. 9(c) and (f)). The photodetection parameters of device-2 and device-3 are summarized in Table S5. Furthermore, the variation in the drain current under different intensities of UV/blue light was calculated for all three devices, showing a linear behavior (Fig. S23).
 |
| | Fig. 9 Photoelectrical behavior of the organic/inorganic bilayer TFTs under different intensities of blue light; (a) photosensitivity (S), (b) photoresponsivity (R) and (c) detectivity (D*) of device-2; (d) photosensitivity (S), (e) photoresponsivity (R) and (f) detectivity (D*) of device-3. | |
Transient photoresponse of the TFTs
The zero-gate-bias transient photoresponse of the TFTs was investigated at a 1-V drain bias under blue-light illumination with a light intensity of 15 W m−2, as shown in Fig. 10. The transient response under UV- and blue-light illumination for device-2 is presented in Fig. 10(a), while Fig. 10(b) shows device-3 under blue-light illumination only. The rise time and decay time were calculated with the help of a single cycle in the transient response curve. Although the rise time and decay time of these phototransistors were ∼100 s, which is quite high, this value could have originated from the low hole mobility of the organic molecules. During hole transport, this inherent behavior of the organic molecules resulted in slow charge-carrier trapping and de-trapping processes, effectively increasing the transient response of the device.
 |
| | Fig. 10 Transient response of the phototransistor for multiple cycles under blue light for (a) device-2 and (b) for device-3. | |
Conclusion
In conclusion, we used a bimodal strategy for successfully establishing the contrasting and comparable photophysical behavior of the D–σ–A and D–π–A systems. We designed and synthesized four compounds, AnBf, CbBf (D–σ–A) and AnBr, CbBr (D–π–A). The variation of the spacers between the D and A moieties from the flexible to rigid spacers facilitated contrasting optical properties; however, the change of the donor strength led to comparable optical behavior. In this context, owing to their flexible construction, the D–σ–A system, although weakly emissive in solution, displayed AIE and a strong solid-state emission. On the other hand, the rigid scaffold of the D–π–A system caused intense emission in dilute solutions along with a typical ACQ effect. AnBf demonstrated strong dual emission, whereas CbBr had a higher quantum yield, indicating carbazole's superior electron-donating ability. Dual emission in AnBf could have been related to excimer formation and the CT-state emission. Furthermore, the rigid and flexible spacers caused intramolecular and intermolecular charge transfer in AnBr/CbBr and AnBf/CbBf, respectively. Theoretical studies further authenticated the experimental data as it was found that the alkyl spacers were not involved in the HOMO–LUMO distribution. Additionally, the HOMO–LUMO gap was found to be smaller in AnBf/CbBf compared to AnBr/CbBr, further suggesting the suppression of the ICT pathways in the former. The unique molecular design of AnBf and CbBf as AIEgens was explored through the fabrication of photodetectors, where CbBf (device-2) and AnBf (device-3) molecules functioned as blue-light-absorbing layers. device-2 exhibited an excellent photoresponsivity relative to device-3, which can be related to the reduced HOMO–LUMO gap of CbBf. Thus, this is the first ever systematic study aimed at predicting the photophysical behavior of D–A molecules by taking into account the two most relevant variables, i.e. the spacer and the relative strength of the donor or acceptor. The resulting properties have been well utilized in organic electronic devices.
Conflicts of interest
The authors declare no competing financial interests.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: 1H, 13C, 11B, 19F NMR, ESI-MS, UV-vis, fluorescence spectra, plots and tables. See DOI: https://doi.org/10.1039/d5tc02873f.
CCDC 2339002 (AnBr) contain the supplementary crystallographic data for this paper.61
Acknowledgements
Authors gratefully acknowledge the Science and Engineering Research Board (SERB), New Delhi, India (Scheme ECR/2018/001810) and BHU IOE cell (Seed/Incentive Grant) for providing financial assistance.
References
- K. W. Lee, Y. Wan, Z. Huang, Q. Zhao, S. Li and C. S. Lee, Adv. Mater., 2024, 36, 2306492 CrossRef CAS PubMed.
- Y. Hu, J. Wang, C. Yan and P. Cheng, Nat. Rev. Mater., 2022, 7, 836–838 CrossRef CAS.
- A. Cardone and A. L. Capodilupo, Materials, 2022, 15, 6333 CrossRef CAS PubMed.
- Y. Lu, W. Sun, J. Du, J. Fan and X. Peng, JACS Au, 2023, 3, 682–699 CrossRef CAS PubMed.
- Y. Liu, H. Wang, F. Ke, X. Ji, N. Chen, K. Zhang, Z. Yang, Y. Chen and Z. Zhang, Adv. Ther., 2024, 7, 2300400 CrossRef CAS.
- L. Sun, J. Wang, B. Yang, X. Wang, G. Yang, X. Wang, Y. Jiang, T. Wang and J. Jiang, RSC Adv., 2021, 11, 10061–10074 RSC.
- N. Kwon, H. Kim, X. Li and J. Yoon, Chem. Sci., 2021, 12, 7248–7268 RSC.
- A. Escudero Belmonte, C. Carrillo Carrión, M. d C. Castillejos Anguiano, E. Romero Ben, C. Rosales Barrios and N. Khiar, Mater. Chem. Front., 2021, 5, 3788–3812 RSC.
- Y. Li, P. Zhang, Y. Xie, J. Yang, Y. Yang, L. Shi, W. Wu and Z. Li, Biomaterials, 2023, 299, 122182 CrossRef CAS.
- H. Zhang, W. Han, J. Han, P. Xu and P. Jiang, J. Mater. Sci., 2022, 57, 14620–14654 CrossRef CAS.
- Z. Wu, A. C. Midgley, D. Kong and D. Ding, Mater. Today Chem., 2022, 17, 100481 CAS.
- E. Arroyo, B. T. Herrero, J. M. de la Fuente, M. Ocaña and A. I. Becerro, Inorg. Chem. Front., 2022, 9, 2454–2461 RSC.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 RSC.
- S. Campagna, F. Nastasi, G. La Ganga, S. Serroni, A. Santoro, A. Arrigo and F. Puntoriero, Phys. Chem. Chem. Phys., 2023, 25, 1504–1512 RSC.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 8355–8364 RSC.
- H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3534–3539 CrossRef CAS PubMed.
- D. N. Beratan, C. Liu, A. Migliore, N. F. Polizzi, S. S. Skourtis, P. Zhang and Y. Zhang, Acc. Chem. Res., 2015, 48, 474–481 CrossRef CAS.
- S. R. Rather and G. D. Scholes, J. Am. Chem. Soc., 2018, 141, 708–722 CrossRef PubMed.
- J. T. Buck, R. W. Wilson and T. Mani, J. Phys. Chem. Lett., 2019, 10, 3080–3086 CrossRef CAS.
- X. Chen, A. A. Sukhanov, M. Taddei, B. Dick, J. Zhao, V. K. Voronkova and M. Di Donato, J. Phys. Chem. Lett., 2022, 13, 8740–8748 CrossRef CAS.
- H. Y. Chung, J. Oh, J.-H. Park, I. Cho, W. S. Yoon, J. E. Kwon, D. Kim and S. Y. Park, J. Phys. Chem. C, 2020, 124, 18502–18512 CrossRef CAS.
- K. Susumu, J. A. Fisher, J. Zheng, D. N. Beratan, A. G. Yodh and M. J. Therien, J. Phys. Chem. A, 2011, 115, 5525–5539 CrossRef CAS.
- A. J. Auty, P. A. Scattergood, T. Keane, T. Cheng, G. Wu, H. Carson, J. Shipp, A. Sadler, T. Roseveare and I. V. Sazanovich, Chem. Sci., 2023, 14, 11417–11428 RSC.
- X. Chen, X. Zhang, X. Xiao, Z. Wang and J. Zhao, Angew. Chem., Int. Ed., 2023, 62, 202216010 CrossRef.
- R. Rajagopalan, S. S. Shankar, N. Balasubramaniyan and G. D. Sharma, ACS Appl. Mater. Interfaces, 2023, 15, 13405–13414 CrossRef CAS.
- T. Han, Y. Wang, J. Xu, N. Zhu, L. Bai, X. Liu, B. Sun, C. Yu, Q. Meng and J. Wang, Chem. Sci., 2022, 13, 13201–13211 RSC.
- X. Wang, Z. Liu, S. Geng, Z. Zhong, H. Li, X. J. Feng, Z. Zhao and H. Lu, J. Mater. Chem. C, 2023, 11, 5316–5323 RSC.
- R. Rybakiewicz-Sekita, P. Toman, R. Ganczarczyk, J. Drapala, P. Ledwon, M. Banasiewicz, L. Skorka, A. Matyjasiak, M. Zagorska and A. Pron, J. Phys. Chem. B, 2022, 126, 4089–4105 CrossRef CAS.
- M. Z. K. Baig, B. Prusti, D. Roy, P. K. Sahu, M. Sarkar, A. Sharma and M. Chakravarty, ACS Omega, 2018, 3, 9114–9125 CrossRef CAS PubMed.
- Y. Huang, Y. Xu, K. Liu, Y. Fu, A. G. Ricciardulli, F. Wang, S. Yang, J. Ma, M. Li, Z. Qian and R. Wang, ACS Mater. Lett., 2024, 6, 1069–1076 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- B. Rudek, S. K. Son, L. Foucar, S. W. Epp, B. Erk, R. Hartmann, M. Adolph, R. Andritschke, A. Aquila, N. Berrah and C. Bostedt, Nat. Photonics, 2012, 6, 858–865 CrossRef CAS.
- C. Wang, X. Zhang and W. Hu, Chem. Soc. Rev., 2020, 49, 653–670 RSC.
- H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158–6183 CrossRef CAS.
- K. Liu, B. Ouyang, X. Guo, Y. Guo and Y. Liu, npj Flexible Electron., 2022, 6, 1 CrossRef CAS.
- R. Liu, W. Yang, W. Xu, J. Deng, C. Ding, Y. Guo, L. Zheng, J. Sun and M. Li, ACS Appl. Polym. Mater., 2022, 4, 2233–2250 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- P. Cheng, X. Du, S. Chen, K. Chen, Y. Yuan, J. Shao, Q. Shen, P. Sun and Q. Fan, ACS Appl. Nano Mater., 2023, 6, 10736–10745 CrossRef CAS.
- K. Debsharma, J. Santhi, B. Baire and E. Prasad, ACS Appl. Mater. Interfaces, 2019, 11, 48249–48260 CrossRef CAS PubMed.
- X. Du, H. Su, L. Zhao, X. Xing, B. Wang, D. Qiu, J. Wang and M.-S. Yuan, Mater. Chem. Front., 2021, 5, 7508–7517 RSC.
- W. Xu, M. M. Lee, Z. Zhang, H. H. Sung, I. D. Williams, R. T. Kwok, J. W. Lam, D. Wang and B. Z. Tang, Chem. Sci., 2019, 10, 3494–3501 RSC.
- G. Li, X. Liu, M. Wu, R. Zeng, Q. Li, A. Yuan and C. Shi, Dyes Pigm., 2023, 208, 110805 CrossRef.
- P.-L. Lu, K. Li, L. Shi, X. Liu, M.-L. Feng, H.-Z. He, H. Yang and X.-Q. Yu, Adv. Mater., 2020, 1, 61–70 RSC.
- G. Dai, M. Zhang, K. Wang, X. Fan, Y. Shi, D. Sun, W. Liu, J. Chen, J. Yu and X. Ou, ACS Appl. Mater. Interfaces, 2021, 13, 25193–25201 CrossRef CAS PubMed.
- T. Wang, Z. Hu, X. Nie, L. Huang, M. Hui, X. Sun and G. Zhang, Nat. Commun., 2021, 12, 1364 CrossRef CAS.
- M. J. Kim, M. Ahn and K. R. Wee, Mater. Adv., 2021, 2, 5371–5380 RSC.
- L. Feng, C. Wang, X. Deng, X. Miao, J. Wang, Y. Wang and Z. Li, Mater. Chem. Front., 2018, 2, 264–269 RSC.
-
D. D. Perrin, W. L. F. Armango and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon, Oxford, UK, 1986 Search PubMed.
-
G. M. Sheldrick, SHELXL-97, Program for X-ray Crystal Structure Refinement, Gottingen University, Germany, 1997 Search PubMed.
-
G. M. Sheldrick, SHELXL-97, Program for X-ray Crystal Structure Solution, Gottingen University, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek and IUCr, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 34 Search PubMed.
- Y. Liu, K. Li, M. Y. Wu, Y. H. Liu, Y. M. Xie and X. Q. Yu, Chem. Commun., 2015, 51, 10236–10239 RSC.
- P. Taranekar, A. Baba, J. Y. Park, T. M. Fulghum and R. Advincula, Adv. Funct. Mater., 2006, 16, 2000–2007 CrossRef CAS.
- H. Ju, T. Hiraoka, H. Horita, E. Lee, M. Ikeda, S. Kuwahara and Y. Habata, Dalton Trans., 2022, 51, 15530–15537 RSC.
- C. Zhao, Z. Ding, Y. Zhang, Z. Ni, S. Li, S. Gong, B. Zou, K. Wang and L. Yu, Chem. Sci., 2023, 14, 1089–1096 RSC.
- B. N. Pal, B. M. Dhar, K. C. See and H. E. Katz, Nat. Mater., 2009, 8, 898–903 CrossRef CAS.
- A. Sharma, N. K. Chourasia, A. Sugathan, Y. Kumar, S. Jit, S.-W. Liu, A. Pandey, S. Biring and B. N. Pal, J. Mater. Chem. C, 2018, 6, 790–798 RSC.
- P. K. Aich, Z. Genene, U. Pandey, A. K. Yadav, E. Wang and B. N. Pal, ACS Photonics, 2024, 11(9), 3704–3712 CrossRef CAS.
-
CCDC 2339002: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2jhxpn.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b and
Roop Shikha
Singh
b and
Roop Shikha
Singh
















