DOI:
10.1039/D5TB01511A
(Paper)
J. Mater. Chem. B, 2026,
14, 3158-3186
Multifaceted antibacterial action of dihydrofurocoumarins against drug-resistant Escherichia coli: biofilm inhibition, membrane disruption, metabolic dysfunction, and oxidative stress damage
Received
25th June 2025
, Accepted 1st December 2025
First published on 6th December 2025
Abstract
The alarming rise in antibiotic resistance necessitates the urgent development of novel therapeutic agents. Herein, we report a bifunctional approach to synthesize two series of dihydrofurocoumarins (DHFCs), one incorporating naphthalimide and the other featuring coumarin analogues, designed to explore their antibacterial potential and ability to combat antibiotic resistance through structural diversification. Preliminary assessments reveal that some synthesized analogues exhibit significant antibacterial potency. Notably, analogues with electron-withdrawing substituents, particularly 16b and 21e (MIC = 1.56 µg mL−1), display outstanding activity against E. coli, demonstrating a higher potency than the marketed antibiotic amoxicillin. The low-frequency resistance observed for analogues 16b and 21e, as evidenced by stable MIC values even after extended passages, may be attributed to their rapid bactericidal action. Additionally, both analogues strongly inhibit biofilm formation, disrupting a critical pathway involved in the development of drug resistance. Mechanistic investigations revealed that both analogues effectively disrupt bacterial membranes, triggering cytoplasmic leakage and a significant loss of metabolic activity. They also induce reactive oxygen species (ROS) generation, catalyzing the oxidation of GSH to GSSG, thereby diminishing cellular GSH activity and weakening the bacterial antioxidant defense system, ultimately leading to oxidative damage and cell death. Active analogues were evaluated for their binding affinity to human serum albumin (HSA), demonstrating a balanced binding profile with optimal binding constants, indicative of their potential to facilitate targeted delivery without compromising drug release at the intended site. Site marker drug displacement studies further identified their binding sites, showing that 16b exhibited a preference for Sudlow site I, while 21e selectively associated with the heme site on HSA. Molecular docking studies further corroborated these findings, revealing perfect alignment with experimental results. Further investigations indicated that both active analogues intercalated into DNA, forming DNA–16b/21e complexes that disrupted essential biological functions, leading to bacterial death. Quantum chemical insights revealed a narrower HOMO–LUMO energy gap, facilitating electronic transitions and enhancing molecular reactivity, which may be pivotal for their antibacterial effectiveness. Amidst the limitations of conventional antibiotics, these findings underscore the potential of dihydrofurocoumarins as potent multitarget, broad-spectrum antibacterial agents. Their ability to impair bacterial defense mechanisms and combat persistent pathogens presents a promising avenue for advancing antibacterial therapeutics, paving the way for further clinical exploration and the development of novel antibacterial analogues.
1. Introduction
Antimicrobial resistance (AMR), with bacterial strains at the forefront, has emerged as a pressing global health problem, severely compromising the effectiveness of both therapeutic and preventive measures against infections, with approximately 5 million deaths in 2019.1 This silent pandemic has not only led to a surge in infections caused by resistant pathogens but also rapidly diminished the resources of effective antibiotics, with projections warning of a potential $100 trillion economic burden by 2050, according to the WHO.2 Among the most concerning threats are ESKAPE pathogens (Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.), which can evade or escape standard therapies via diverse antimicrobial-resistance mechanisms and biofilm formation and are considered highly virulent agents causing life-threatening infections, thereby necessitating the development of novel antibacterial drug candidates.3 The spread of resistance is further worsened by antibiotic pollution, as unmetabolized drugs are excreted into the environment, facilitating the spread of resistance beyond humans and animals. Antibiotics enter the environment through human excretion, livestock farming, urban wastewater, agricultural activities, improper disposal by healthcare professionals and patients, and primarily due to their indiscriminate use. Consequently, environmental antibiotic buildup pressures bacteria to adapt, leading to resistance traits that fuel the rise of antimicrobial resistance.4 This escalating challenge has highlighted the limitations of traditional antibiotic research, which predominantly focused on refining and modifying established drug scaffolds to create new therapeutics. However, after decades of iterative optimization around the same core chemical structures, such approaches are now insufficient to keep up with the accelerating emergence and spread of antimicrobial resistance.5,6 As a result, researchers worldwide are intensifying efforts to develop next-generation antimicrobial agents, designed to overcome resistance challenges through novel molecular structures and diverse mechanisms that surpass the limitations of traditional antibiotics.
A wide range of structurally diverse heterocyclic derivatives have been explored for their potential to address multidrug-resistant bacterial infections, offering several mechanisms of action.7 An analysis of drug approvals in 2021 revealed that nine of the twelve molecules endorsed by the US FDA possessed heterocyclic scaffolds as essential pharmacophoric elements. Incorporation of these scaffolds into drug candidates enables fine-tuning of their physicochemical attributes, absorption and distribution patterns, therapeutic efficacy, and safety parameters.8 In this work, the strategic design of a target molecule incorporating heterocyclic bioactive rings for the development of antibacterial agents is driven by the following key considerations:
• Coumarin constitutes a highly valuable and biologically significant heterocyclic scaffold, prevalent in a multitude of therapeutic frameworks.9 Owing to its planar aromatic framework containing a lactone ring, coumarin can engage in strong non-covalent interactions with various biological targets, including DNA, thereby serving as an effective pharmacophore for rational drug design and exhibiting a broad spectrum of pharmacological activities.10–12 The antibacterial potential of coumarins is particularly noteworthy, primarily attributed to their ability to inhibit key enzymes involved in fatty acid biosynthesis, thereby disrupting essential bacterial metabolic processes.13 Prominent aminocoumarin antibiotics, such as novobiocin, chlorobiocin, and coumermycin A1, target bacterial DNA gyrase, contributing significantly to their antibacterial efficacy.14 Although these antibiotics have shown issues related to toxicity, resistance, and adverse effects, the core coumarin scaffold continues to inspire the design of novel antibacterial agents with improved efficacy and safety profiles.15 These features collectively justify the strategic incorporation of coumarin in the development of new antibacterial agents.
• Furocoumarins represent a distinguished class of naturally occurring bioactive analogues, widely recognized for their applications in traditional remedies and modern clinically approved drugs.16 Building upon this structural motif, trans-2,3-dihydrofuro[3,2-c]coumarins (DHFCs), belonging to the class of oxa-heterocycles, have gathered considerable interest, which imparts profound medicinal significance and supports their use in a variety of therapeutic formulations and in a wide array of pharmaceutical agents.17,18
• Bis-coumarins are seldom present in nature, mostly existing as dimers within select plant species. Their unique biological activities have recently sparked increased scientific interest.19 A wide spectrum of pharmacological activities have been attributed to bis-coumarins, including enzyme inhibition, anticoagulant and hemorrhagic effects, as well as antifungal, anti-helminthic, anti-tumor, anti-HIV, anti-apoptotic, antioxidant, and analgesic activities.20,21 Therefore, the bis-coumarin framework, containing two pharmacophoric units, is thought to enhance antibacterial activity through possible synergistic interactions and may offer advantages in overcoming bacterial resistance by improving target binding and prolonging biological effects.
• Within the realm of heterocyclic frameworks, naphthalimide has emerged as a compelling scaffold in the design of antibacterial agents, by virtue of its multitargeting potential and its capacity to embody innovative modes of action, thus steadily gaining momentum as a promising antibacterial candidate in recent times.22 Its inherent hydrophobicity facilitates effective integration into the phospholipid bilayer of bacterial membranes, thereby compromising membrane integrity and inducing cytoplasmic leakage, which eventually leads to cell death. This unique mode of action positions the naphthalimide core as a privileged structural motif for synthesizing membrane-targeting antibacterial candidates capable of addressing the growing challenge of cross-resistance.23
Recognizing the inherent biological promise of aforementioned heterocycles, a new generation of antimicrobial agents were synthesized through the strategic integration of these pharmacologically active moieties. The molecular hybridization approach strengthens target engagement, increases therapeutic efficacy, reduces toxicity, addresses resistance, and introduces entirely new biological functions.24 In line with this approach, a series of dihydrofurocoumarins were synthesized using a bifunctional annulation strategy, resulting in two structurally distinct classes of analogues. In these hybrids, the C-2 position of the dihydrofurocoumarin core served as a crucial linkage site, connecting to the naphthalimide and coumarin moieties via amide and carbonyl linkers, respectively, thereby generating two distinct structural frameworks with potential therapeutic relevance (Fig. 1). To evaluate the effect of various substituents, a series of analogues were synthesized by introducing different aromatic rings at the C-3 position of the dihydrofurocoumarin core, along with morpholine and aryl groups at the C-4 position of the naphthalimide scaffold. Each of these synthesized analogues was assessed for antibacterial activity against a panel of eight Gram-positive and Gram-negative strains. The results revealed broad-spectrum potential, with notable inhibition observed across multiple bacterial species. Among these, Escherichia coli, a Gram-negative, rod-shaped bacterium well known in human ecology as the first bacterial species to colonize after birth, was selected for further investigation as it exhibited the highest susceptibility to most of the synthesized analogues and is also clinically significant due to its increasing resistance to current antibiotics, which has led to its inclusion, along with other members of the Enterobacteriaceae family, in the World Health Organization (WHO) ‘priority pathogens’ list.25,26 This usually benign commensal can readily transform into a remarkably adaptable pathogen responsible for severe illnesses, including gastroenteritis and extraintestinal infections affecting the urinary tract, bloodstream, and central nervous system.27 It has been recognized as a principal indicator of antibiotic-resistant bacteria and clinically important resistance genes, attributable to its widespread occurrence, clinical relevance, and ability to acquire conjugative plasmids.28 Globally, E. coli is estimated to cause approximately 150 million cases of uncomplicated cystitis each year and over two million deaths resulting from infant diarrhea and extraintestinal infections. Moreover, its cell wall, composed of a thin peptidoglycan layer surrounded by an outer membrane, forms a natural barrier that restricts the entry of antibiotics such as penicillin, contributing to its resilience and clinical challenges.29 Despite extensive efforts to discover new antimicrobials targeting Gram-negative bacteria, no novel antibiotic class has been approved in the last five decades, and most available antibiotics are based on conventional scaffolds, making the emergence of resistance to these drugs readily anticipated. Consequently, there is an urgent need to develop antibiotics with new mechanisms of action to stay ahead in fight against resistance.30 In light of this, our study undertook a detailed investigation of most potent analogues, 16b and 21e, specifically against E. coli, to gain deeper insights into their antimicrobial efficacy and mechanism of action. A comprehensive series of biological assays were performed, including antibiofilm activity, time-kill kinetics, resistance development, membrane permeabilization, metabolic inactivation, oxidative stress induction, and cytotoxicity assessment. Additionally, in silico ADME profiling and quantum chemical studies were carried out to evaluate their pharmacokinetic and electronic characteristics. To further assess their transport potential, binding interactions with human serum albumin (HSA) were examined using spectroscopic and molecular docking studies.
 |
| | Fig. 1 Molecular design of dihydrofurocoumarins as antibacterial agents. | |
2. Results and discussion
2.1. Chemistry
The target dihydrofurocoumarins, along with the envisioned intermediates, were synthesized following the synthetic routes outlined in Schemes 1–8, employing a one-pot multicomponent annulation reaction as the key step. This reaction involved the Knoevenagel condensation of 4-hydroxycoumarin with various aldehydes, followed by Michael-type addition of an aza-ylide, generated through nucleophilic addition of pyridine to bromoacetyl derivatives of naphthalimide/coumarin. The resulting zwitterion then rearranged into its enolate form, which underwent intramolecular cyclization via the SN2 mechanism, leading to the formation of dihydrofurocoumarin analogues. This strategic assembly of key precursors in pyridine under transition metal-free conditions efficiently yielded the desired analogues. Oxidation of readily accessible acenaphthene 1 with potassium dichromate in refluxing acetic acid resulted in formation of 2. Amidation of naphthalic anhydride 2 upon reaction with hydrazine hydrate in ethanol at room temperature led to the formation of 3, which was then subjected to nucleophilic substitution with bromoacetyl bromide to form 4 in 89% yield (Scheme 1). The coupling of 4 and 4-hydroxycoumarin 5 with different aldehydes 6a–f in pyridine at 80 °C for 5–6 h produced the desired naphthalimide tethered dihydrofurocoumarin analogue 7a–f (Scheme 2). 1H NMR of 7a showed doublets at δ 4.88 ppm and δ 5.71 ppm of two aliphatic protons of the furan moiety, 1H singlet of NH at δ 11.46 ppm and the chemical shifts of 8.58–7.33 ppm correspond to the aromatic protons of phenyl, coumarin, and naphthalimide moieties. The 13C NMR spectrum revealed signals for the carbonyl carbons of naphthalimide, amide, and coumarin, all observed in the range of 167.5–154.7 ppm.
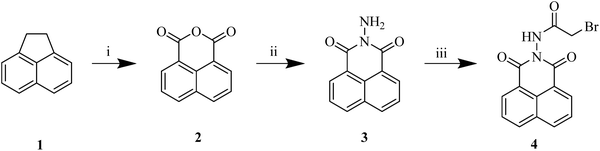 |
| | Scheme 1 Synthesis of 2-bromo-N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (4)a. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reagents and conditions: (i) K2Cr2O7, acetic acid, reflux, 3 h, 95%; (ii) hydrazine hydrate, ethanol, rt, 30 min, 84%; (iii) bromoacetyl bromide, 1,2-dichloroethane, reflux, 10 min, 89%. Reagents and conditions: (i) K2Cr2O7, acetic acid, reflux, 3 h, 95%; (ii) hydrazine hydrate, ethanol, rt, 30 min, 84%; (iii) bromoacetyl bromide, 1,2-dichloroethane, reflux, 10 min, 89%. | |
 |
| | Scheme 2 Synthesis of naphthalimide tethered dihydrofurocoumarins (7a–f).a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reagents and conditions: (i) pyridine, 80 °C, 5–6 h, 65–88%. Reagents and conditions: (i) pyridine, 80 °C, 5–6 h, 65–88%. | |
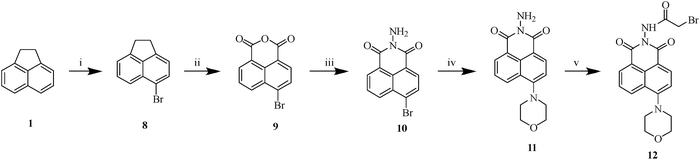 |
| | Scheme 3 Synthesis of 2-bromo-N-(6-morpholino-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (12).a aReagents and conditions: (i) NBS, DMF, rt, 3 h, 95%; (ii) Na2Cr2O7·2H2O, acetic acid, reflux, 4 h, 82%; (iii) hydrazine hydrate, ethanol, rt, 30 min, 84%; (iv) morpholine, K2CO3, DMF, 100 °C, 3 h; (v) bromoacetyl bromide, 1,2-dichloroethane, reflux, 10 min, 85%. | |
 |
| | Scheme 4 Synthesis of 6-amino substituted naphthalimide tethered dihydrofurocoumarin (13).a aReagents and conditions: (i) pyridine, 80 °C, 5 h, 69%. | |
 |
| | Scheme 5 Synthesis of 6-aryl substituted 2-bromo-N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (15a–f)a. aReagents and conditions: (i) RB(OH)2, Pd(PPh3)4, K2CO3, CH3CN:H2O (9:1), N2, reflux, 3–4 h; (ii) bromoacetyl bromide, 1,2-dichloroethane, reflux, 10 min, 79–88%. | |
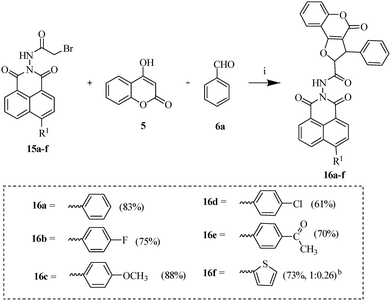 |
| | Scheme 6 Synthesis of 6-aryl substituted naphthalimide tethered dihydrofurocoumarins (16a–f).a aReagents and conditions: (i) pyridine, 80 °C, 7–8 h, 61–88%. bThe major diastereomers were determined by 1H NMR analysis. | |
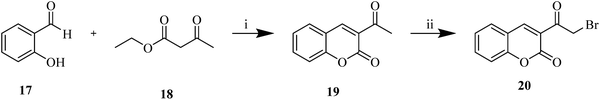 |
| | Scheme 7 Synthesis of 3-(2-bromoacetyl)-2H-chromen-2-one (20).a aReagents and conditions: (i) piperidine, rt, 10 min, 87%; (ii) NBS, p-TSA·H2O, CHCl3/CH3CN (5/1), reflux, 12 h, 95%. | |
 |
| | Scheme 8 Synthesis of coumarin tethered dihydrofurocoumarin yielding bis-coumarins (21a–l).a aReagents and conditions: (i) pyridine, 80 °C, 6–7 h, 41–95%. bThe major diastereomers were determined by 1H NMR analysis. | |
To explore the influence of diverse substituents on antibacterial efficacy, morpholine and various aromatic rings were functionalized at the C-4 position of naphthalimide. Analogue 11 was synthesized by reacting key precursor 10 with morpholine in the presence of potassium carbonate in heated DMF for 3 h. Similarly, derivatives 14a–f were obtained via the Suzuki–Miyaura cross-coupling reaction using various boronic acids, potassium carbonate, and tetrakis(triphenylphosphine)palladium(0) in a refluxing acetonitrile–water mixture (9:1) under an inert atmosphere for 3–4 h. Subsequently, intermediates 12 and 15a–f were synthesized by treating respective 11 and 14a–f with bromoacetyl bromide in refluxing 1,2-dichloroethane, affording good yields (Schemes 3 and 5). Further reaction of these intermediates with 4-hydroxycoumarin 5 and benzaldehyde 6a in pyridine yielded the final analogues 13 (69%) and 16a–f (61–88%), respectively (Schemes 4 and 6).
To assess the potency and extend the substrate versatility of the aforementioned protocol, unsymmetrical bis-coumarins (21a–l) were likewise synthesized through a sequential approach. Initially, 3-acetyl-2H-chromen-2-one 19 was subjected to bromination to afford the key intermediate, 3-(bromoacetyl)coumarin 20 (Scheme 7), which was subsequently reacted with hydroxycoumarin 5 and various aldehydes (6a–l) to yield the final analogues 21a–l (Scheme 8). 1H NMR, 13C NMR, and HRMS were used for characterization of synthesized analogues (Fig. S1–S66). All dihydrofurocoumarin analogues were obtained in moderate to excellent yields ranging from 41% to 95%. Notably, the synthesis of analogues 7a–f, 21a, and 21h circumvented the necessity for column chromatography, as the crude products were efficiently purified through solvent washing to eliminate residual impurities, if any. Overall, this operationally streamlined synthesis obviates the need for intermediate purification, enabling gram-scale reactions with optimal efficiency, minimal reaction time, and high atom economy while maintaining a multicomponent framework and proceeding under mild reaction conditions, thereby offering a practical and versatile framework for diverse synthetic and biological applications.
2.2. Antibacterial activity
An investigation into the antibacterial efficacy of dihydrofurocoumarin analogues was conducted in vitro using eight selected bacterial strains, including Gram positive (Staphylococcus aureus-902, Enterococcus faecalis-6845, Listeria. species-4214, Bacillus subtilis-441) and Gram negative (Serratia marcescens-2645, Escherichia coli-448, Salmonella enterica-1165, Acinetobacter calcoaceticus-1948) bacteria, with minimum inhibitory concentrations (MICs, µg mL−1) serving as the primary metric (Table 1). Evaluation of antibacterial properties revealed insightful structure–activity correlations, as certain synthesized analogues showed pronounced antibacterial efficacy, reflecting the substantial impact of molecular substituents. The comparative potency of these analogues was assessed with amoxicillin and tetracycline as reference antibiotics.
Table 1
In vitro antibacterial data (MICs, µg mL−1) for analogues 7a–f, 13, 16a–f and 21a–l
Among the series of naphthalimide-based analogues (7a–f), considerable antibacterial activity was observed against the tested strains. The variation in activity was influenced by the electronic characteristics of the phenyl group introduced at the C-2 position of the dihydrofurocoumarin core. Notably, analogues with electron-donating substituents at this position demonstrated enhanced antibacterial potency compared to those with electron-withdrawing groups. Among these, 7b and 7d, bearing para-substituted methyl and methoxy groups, exhibited particularly strong activity against Gram-positive strain, S. aureus, with MIC values of 3.12 µg mL−1 and 1.56 µg mL−1, respectively, highlighting the favorable impact of electron-donating substituents. Further structural optimization was performed to investigate the impact on bioactivity by introducing secondary amine 13 and various aryl rings (16a–f) at the C-4 position of naphthalimide. Notably, analogue 13, incorporating morpholine at the C-4 position, demonstrated remarkable broad-spectrum antibacterial activity, with particularly potent effects against S. aureus, E. faecalis, and E. coli, yielding an MIC of 1.56 µg mL−1. This potency may arise from the inclusion of morpholine, a basic group that facilitates brain permeability, while reinforcing molecular interactions at the target site. Moreover, it is also believed to modulate pharmacokinetic and pharmacodynamic properties, potentially enhancing overall efficacy and contributing to antibacterial activity.31 Notably, the presence of morpholine in finafloxacin, an FDA-approved antibacterial agent, further underscores its significance in the design of biologically active molecules.32 On the other hand, phenyl rings bearing electron-withdrawing substituents at the C-4 position of naphthalimide generally exhibited superior antibacterial activity compared to those with electron-donating groups. Among these, analogue 16b, bearing a fluoro substituent, effectively inhibited the growth of most tested bacterial strains, showing a low MIC values of 1.56 µg mL−1, except for E. faecalis (MIC = 6.25 µg mL−1), L. species (MIC = 3.12 µg mL−1), and A. calcoaceticus (MIC = 3.12 µg mL−1). The beneficial role of fluorine in 16b may parallel its established impact in fluoroquinolone antibiotics such as norfloxacin and ciprofloxacin, where it enhances antibacterial efficacy by increasing tissue permeability.33 This effect is well-documented in enoxacin, an antibacterial agent, where the introduction of fluorine at the C-6 position increases lipophilicity, enhancing cell penetration, binding affinity, and improving gyrase-complex binding, which in turn boosts gyrase activity.34 Furthermore, the effect of the fluorine atom is evident in antimicrobial quinolones, where it improves bioavailability, potency, and tissue penetration, while exhibiting low toxicity and broadening the spectrum of antimicrobial activity.35 Meanwhile, 16d containing a chloro substituent showed moderate activity across all strains. Notably, 16e, which possesses an acetyl group, demonstrated enhanced activity specifically against E. coli and A. calcoaceticus (MIC = 1.56 µg mL−1). Furthermore, 16f, featuring a thiophene ring, showed pronounced activity against Gram-negative bacteria, particularly E. coli (MIC = 1.56 µg mL−1), S. marcescens and A. calcoaceticus (MIC = 3.12 µg mL−1), but exhibited reduced efficacy against Gram-positive strains. In contrast, 16c with an electron-donating methoxy group displayed no detectable antibacterial activity against any of the tested strains, reinforcing the trend that electron-withdrawing substituents at naphthalimide or electron-donating substituents at dihydrofurocoumarin are crucial for potent antibacterial effects.
Extending these findings to the unsymmetrical bis-coumarin series (21a–l), where the dihydrofurocoumarin scaffold serves as the central motif, substitution at its C-2 position with a phenyl ring, particularly when further functionalized with various substituents, proves crucial for enhancing antibacterial potency. Almost no tested bacteria are sensitive to 21a, which bears an unsubstituted phenyl ring at the C-2 position. However, the addition of electronic substituents on the phenyl ring resulted in notable increase in bacterial efficacy. Analogue 21b, bearing a methyl group, exhibited an MIC of 6.25 µg mL−1 against S. aureus. Further substitution with a chlorine in 21c enhanced activity against Gram-negative strains, with E. coli showing the highest sensitivity (MIC = 1.56 µg mL−1). However, the para-chloro substituted analogue 21d exhibited an MIC of 12.5 µg mL−1 against both S. marcescens and E. coli. Analogue 21e, bearing a nitro group at the meta-position of the phenyl ring, exhibited broad spectrum antibacterial activity, with MIC values ranging from 1.56 to 12.5 µg mL−1. This potent effect may be attributed to the strong electron withdrawing nature of the nitro group, which is known to enhance pharmacological activity in several drugs, such as nitroimidazole, fexinidazole, and ranitidine. Upon entering bacterial cells, the nitro group underwent enzymatic reduction, leading to generation of reactive oxygen species (ROS). These ROS may disrupt the glutathione (GSH)/oxidized glutathione (GSSG) balance, thereby impairing the function of key cellular enzymes and contributing to antibacterial action.36 In contrast, para-nitro substituted analogue 21f exhibited loss of activity against Gram positive bacteria, although it retained moderate efficacy against Gram negative strains. These observations further reinforce the importance of substituent positioning in modulating antibacterial activity. Analogue 21h, featuring a methoxy group at the para-position, demonstrated good activity against S. aureus with an MIC of 3.12 µg mL−1. Additionally, the introduction of a cyano group in 21i proved beneficial, as it exhibited strong activity against all tested strains, particularly S. aureus, E. coli, and S. enterica, with an MIC of 1.56 µg mL−1. The cyano group can be metabolized by organisms without generating toxic cyanide, making it an important modification in drug design, as exemplified by pradofloxacin, a broad-spectrum veterinary antibacterial drug, and azoxystrobin, an antifungal drug.37 The dichloro analogue 21k displayed effective activity against A. calcoacetius with an MIC of 3.12 µg mL−1, while the bromo-substituted analogue 21l showed significantly reduced activity against almost all bacterial strains. Overall, the naphthalimide-containing 16b and the bis-coumarin 21e analogues exhibited the strongest antibacterial activity across nearly all tested strains. Owing to their potent efficacy, both were selected as representative molecules for further biological investigations.
Furthermore, subsequent evaluations were conducted to determine the antibacterial potential of the most active analogues against a panel of interacting bacterial species. To assess polymicrobial interactions, E. coli was co-cultured with: (i) a Gram-positive group comprising S. aureus, E. faecalis, L. species, and B. subtilis (Set 1); (ii) a Gram-negative group including S. marcescens, S. enterica, and A. calcoaceticus (Set 2); and (iii) a mixed group containing all listed strains (Set 3). Antibacterial efficacy of the analogues was assessed against all three bacterial sets using a two-fold serial dilution assay involving multiple interacting species. As shown in Table 2, analogues 16b and 21e exhibited strong antibacterial activity against multiple interacting bacterial species, with MIC values ranging from 1.56 to 12.5 µg mL−1, highlighting their potential as effective antimicrobial agents for the treatment of polymicrobial infections.
Table 2
In vitro antibacterial activity as MIC (µg mL−1) for analogues 16b and 21e against multiple interacting bacterial species
2.3.
In silico ADME predictions
To assess the pharmacokinetic properties and drug-likeness of the active analogues, analysis was performed using SwissADME (Table 3).38 The evaluation adhered to Lipinski's Rule of Five, ensuring compliance with key physicochemical parameters associated with oral bioavailability: MW ≤ 500, RB ≤ 10, HBD ≤ 5, HBA ≤ 10, and logP ≤ 5.39 The analysis indicates that the active analogue 21e complies with Lipinski's Rule of Five without violation. In contrast, the molecular weight and log P of 16b highlight aspects that may benefit from further optimization. With a total polar surface area (TPSA) below 140 Å, both analogues demonstrated improved drug permeability across cellular plasma membranes and enhanced oral bioavailability, unlike amoxicillin and tetracycline, which exceed this threshold.40,41 The inability of both analogues to cross the blood–brain barrier (BBB) suggests a reduced risk of neurotoxicity comparable to that of the reference drugs. Furthermore, neither analogue is a substrate for P-glycoprotein (P-gp), indicating minimal interaction with the efflux transporter and potentially reducing the likelihood of treatment resistance.42,43 Notably, none of the active analogues exhibited PAINS alerts. The observed results indicate that both analogues share comparable drug-likeness profiles with control drugs, reinforcing their potential for further development as antibacterial agents.
Table 3 ADME evaluation data of analogues 16b, 21e, amoxicillin and tetracyclinea
| Parameters |
16b
|
21e
|
Amoxicillin |
Tetracycline |
|
MW: molecular weight, logP: lipophilicity, HBA: hydrogen bond acceptor, HBD: hydrogen bond donor, RB: rotational bonds, BBB: blood–brain barrier, TPSA: topological polar surface area, % Abs: 109–0.345 (TPSA), P-gp: poly-glycoprotein substrate, GI: gastrointestinal absorption, PAINS: Pan Assay Interference Compounds.
|
| MW (g mol−1) |
596.56 |
481.41 |
365.40 |
444.43 |
| logP |
5.63 |
3.57 |
−0.39 |
−0.47 |
| HBA |
7 |
8 |
6 |
9 |
| HBD |
1 |
0 |
4 |
6 |
| RB |
5 |
4 |
5 |
2 |
| Lipinski violations |
2 |
0 |
0 |
1 |
| BBB permeant |
No |
No |
No |
No |
| Bioavailability score |
0.17 |
0.55 |
0.55 |
0.11 |
| TPSA |
107.61 |
132.54 |
158.26 |
181.62 |
| %Abs |
71.87 |
63.27 |
54.40 |
46.34 |
| P-gp substrate |
No |
No |
No |
Yes |
| GI absorption |
Low |
Low |
Low |
Low |
| PAINS |
0 |
0 |
0 |
0 |
2.4. Biofilm inhibition assay
Self-secreted extracellular macromolecules, such as polysaccharides, lipids, extracellular DNA, and proteins, form the foundational matrix of bacterial biofilms. These biofilms significantly enhance bacterial defense against antibiotics, boosting resistance up to 1000-fold compared to their planktonic counterparts.44 They shield bacteria from both the host immune response and the action of antimicrobial agents, resulting in elevated antibiotic resistance and persistent infections often associated with increased levels of inflammatory markers.45 Therefore, targeting biofilm inhibition has emerged as a promising approach within broad-spectrum anti-infective strategies aimed at challenging infections caused by drug-resistant bacterial strains.46 Crystal violet staining was used to assess the biofilm inhibitory activity of analogues 16b and 21e toward E. coli. An increase in concentration (1× MIC, 2× MIC, 4× MIC, and 8× MIC) correlates with rise in the biofilm inhibition capacity of both analogues 16b and 21e, which are capable of inhibiting E. coli biofilm formation by 45.7% and 37.9% at their 1× MIC values, with inhibition increasing to 86.5% and 79.3%, respectively, at 8× MIC, with the former showing a greater increase in inhibition compared to the latter (Fig. 2a). These observations point to the efficacy of both analogues in suppressing E. coli biofilm formation, which may help hinder the development of bacterial resistance over time.
 |
| | Fig. 2 (a) Inhibition of biofilms; time-killing kinetics of E. coli by (b) 16b and (c) 21e at various concentrations; (d) bacterial susceptibility evaluation of 16b, 21e, and tetracycline toward E. coli; The experiments were performed in triplicate; *p < 0.05; **p < 0.01; ***p < 0.001; n = 3. | |
2.5. Kinetics of bactericidal activity
Rapid bactericidal action is a distinguishing trait of effective antibacterial agents, enabling swift recovery and limiting the emergence of resistance.47 Hence, the time-kill kinetic study is performed for determining the analogue's bactericidal potential at varying concentrations, as confirmed by its effectiveness against E. coli during distinct exponential growth stages. The control inoculum of E. coli progressed into its exponential phase within 3 hours, but the active analogues effectively stifled its growth and induced bacterial cell death. At its 1× MIC, analogue 16b killed the bacteria within 3 h, while 21e required 5 h to do the same. Increasing the concentration to 2× MIC, 4× MIC, and 8× MIC significantly accelerated the bactericidal action, reducing the time to eliminate the bacteria to 1.5 h for 16b and 3.5 h for 21e (Fig. 2b and c). Therefore, the substantial dose-dependent bactericidal activity of the analogues against E. coli highlights their potential to reduce the emergence of resistance.
2.6. Bacterial susceptibility evaluation
Bacterial resistance undermines the efficacy of clinical drugs, often resulting in treatment failure and substantially hindering the ability to manage infections.48 To determine drug resistance, changes in MICs were assessed for analogues 16b and 21e over 20 consecutive passages against E. coli, with tetracycline serving as the positive control (Fig. 2d). These analogues demonstrated enhanced and long-lasting inhibitory activity against the tested strain, outperforming the control drug tetracycline, while maintaining consistent MIC values throughout the study, emphasizing their sustained effectiveness. In contrast, a marked increase in MIC values of tetracycline was observed after 8 passages. Therefore, the minimal tendency of these analogues to induce drug resistance against E. coli positions them as promising candidates capable of addressing the growing challenge of multi-drug resistant infections.
2.7. Membrane permeability
The bacterial membrane serves as a vital line of defense, playing a key role in preserving intracellular stability and supporting organized biochemical processes.49 The cell membrane is regarded as a crucial target in the development of antibacterial agents, primarily because membrane-disrupting mechanisms pose a lower risk of target mutations, which translates to a reduced likelihood of developing drug resistance.50 Therefore, the membrane-disruptive effects of dihydrofurocoumarin analogues 16b and 21e were investigated by assessing outer membrane permeability, inner membrane permeabilization, cytoplasmic membrane depolarization, nucleic acid leakage and morphological alterations in E. coli cells.
2.7.1. Outer membrane permeability.
Due to a specialized outer membrane predominantly made of amphiphilic lipopolysaccharides, Gram-negative bacteria are less penetrable to hydrophobic molecules than Gram-positive bacteria and these amphiphilic lipopolysaccharides play a critical role in bacterial drug resistance, cellular adhesion, invasion, and biofilm formation. To assess outer membrane permeabilization in E. coli, the fluorescent dye 1-N-phenylnaphthylamine (NPN) was utilized.51 Treatment with 16b and 21e led to a steady increase in fluorescence intensity in E. coli at all concentrations (1 to 8× MIC), reflecting damage to the bacterial outer membrane (Fig. 3a). These observations suggest that both analogues may interact with the outer membrane of E. coli, potentially influencing permeability and contributing to their antibacterial action, which could help mitigate resistance development.
 |
| | Fig. 3 (a) Outer membrane and (b) inner membrane permeability of E. coli treated with 16b and 21e in a concentration dependent manner; *p < 0.05; **p < 0.01; ***p < 0.001; n = 3. | |
2.7.2. Inner membrane permeability.
The low permeability of antibacterial agents is primarily attributed to the protective barrier of bacterial cell membranes. To further investigate this, a propidium iodide (PI) assay was employed to evaluate inner membrane permeability in E. coli upon treatment with increasing concentrations (1 to 8× MIC) of analogues 16b and 21e, using a constant amount of PI.52 The fluorescence intensity of PI was increased significantly with both analogues with increasing concentration relative to untreated bacterial cells. A more pronounced fluorescence response was observed with naphthalimide containing 16b than with 21e, reflecting enhanced inner membrane permeability (Fig. 3b). Therefore, it may be inferred that the active analogues possess the ability to effectively traverse the outer and inner membranes and promote significant bacterial cell damage.
To further elucidate the antimicrobial mechanism, E. coli cells with or without exposure to analogues 16b and 21e were stained with propidium iodide (PI) and examined using confocal laser scanning microscopy (CLSM). Notably, propidium iodide (PI) is a frequently utilized fluorescent turn-on probe that emits red fluorescence upon binding to nucleic acids, allowing the detection of dead bacteria with disrupted cell membranes.53 As shown in Fig. 4, the untreated control group (a) exhibited no detectable fluorescence, whereas E. coli treated with the active analogues 16b (b) and 21e (c) displayed intense red fluorescence, further confirming that both analogues compromise the integrity of the bacterial cell membrane.
 |
| | Fig. 4 Fluorescence micrographs of propidium iodide staining: (a) control and after treatment of E. coli with analogues (b) 16b and (c) 21e at 1.56 µg mL−1 (scale bar: 10 µm). | |
2.7.3. Leakage of cytoplasmic contents.
Once the integrity of the bacterial membrane is irreversibly lost, it causes the efflux of nucleic acids and proteins from the cell. The concentration of nucleic acids leaked from E. coli cells was determined by measuring the UV absorption at 260 nm.54 As shown in Fig. 5, the absorption intensity increases quickly in a concentration-dependent manner for both analogues, verifying the membrane-damaging action of both analogues.
 |
| | Fig. 5 DNA leakage from E. coli in the presence of 16b and 21e at different concentrations; *p < 0.05; **p < 0.01; ***p < 0.001; n = 3. | |
2.7.4. Changes in cell morphology.
The disruption of the E. coli membrane by active analogues was further substantiated by alterations in bacterial morphology, as revealed through scanning electron microscopy (SEM) analysis. As shown in Fig. 6a, SEM images of untreated E. coli depict well-defined, symmetrical rod-shaped cells with preserved structural integrity. Upon treatment with active analogues 16b and 21e at their 1× MIC values, the bacterial cells exhibited ruptured membranes, cytoplasmic leakage and a shrunken, broken appearance (Fig. 6b, c and Fig. S67). Thus, the SEM analysis offers additional support, confirming that both active analogues are effective in disrupting bacterial cell membranes.
 |
| | Fig. 6 SEM images of (a) untreated E. coli and treated E. coli with analogues (b) 16b and (c) 21e at 1× MIC. Scale bar: 5.0 µm. | |
2.8. Beyond primary target inhibition: metabolism and intracellular oxidative stress
To elucidate the mechanism underlying the observed antibacterial activity of analogues 16b and 21e against E. coli, metabolic activity and intracellular oxidative stress were evaluated by quantifying reactive oxygen species, measuring glutathione levels, and analyzing lipid peroxidation. Collectively, these analyses provide insights into bacterial viability, as well as the extent of oxidative damage and redox imbalance induced within the cells.
2.8.1. Measurement of metabolic activity.
The metabolic activity of bacteria is fundamentally linked to their growth and proliferation, predominantly occurring at the cell membrane.55 To monitor metabolic changes in treated E. coli cells, Alamar Blue (resazurin), a redox indicator, was used. In its oxidized state, resazurin is non-fluorescent and appears purple-blue, transitioning to pink or red fluorescence upon reduction to resorufin.56,57 Incubation of E. coli with analogues resulted in significant decrease in metabolic activity, showing a negative correlation with concentration (Fig. 7a). At 8× MIC, the metabolic activity decreased to 23.8% and 36.5% in the presence of analogues 16b and 21e, respectively. Consequently, the incubation of both active analogues with bacterial cells led to membrane destabilization, significantly impairing metabolic efficiency.
 |
| | Fig. 7 (a) Metabolic activity; (b) intracellular ROS production; (c) loss in GSH activity and (d) malondialdehyde (MDA) production in E. coli treated with 16b and 21e. *p < 0.05; **p < 0.01; ***p < 0.001; n = 3. | |
2.8.2. Evaluation of intracellular reactive oxygen species (ROS).
An increase in cellular ROS may amplify antibiotic effectiveness while curbing resistance development, irrespective of specific drug–target interactions.58 Intracellular ROS levels in E. coli treated with active analogues were assessed using the non-fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA), which is oxidized by ROS within cells to form the green-fluorescent 2′,7′-dichlorofluorescein (DCF), indicating ROS generation.59 A concentration-dependent enhancement of emission intensity was evident in E. coli treated with 16b and 21e (Fig. 7b). Thus, both analogues elevate ROS production, driving oxidative damage in E. coli and leading to the ultimate demise of bacterial cells.
To further elucidate the nature of ROS generated by 16b and 21e, their potential to produce singlet oxygen under irradiation was evaluated using ABDA scavenging assay, where 9,10-anthracenediylbis(methylene) dimalonic acid (ABDA) served as the singlet oxygen scavenger. Solutions of ABDA in the presence of 16b and 21e were irradiated and their absorption was monitored by UV-vis spectroscopy. No significant quenching of ABDA absorption was observed, indicating negligible singlet oxygen generation (Fig. S68). This suggests that the ROS produced by 16b and 21e are likely formed via a Type I (electron-transfer) pathway rather than the Type II (energy-transfer) process that typically yields singlet oxygen.60 Consequently, the antibacterial effects of active analogues may proceed through Type I ROS involving superoxide anions (O2−˙) and hydroxyl radicals (˙OH), enhancing oxidative stress and contributing to bacterial cell death.
2.8.3. Determination of glutathione activity.
The evaluation of glutathione served as a supplementary indicator to affirm the ROS levels within the cells. Glutathione (GSH) functions as a key antioxidant in bacteria, critically involved in mitigating oxidative stress and preserving cellular redox homeostasis. External stimuli that induce a surge in ROS levels may oxidize GSH to GSSG, depleting its availability and diminishing its protective antioxidant function. This reduction in antioxidant capacity leaves essential macromolecules vulnerable to oxidative damage, which may contribute to bacterial death.61 Thus, elevated ROS levels lead to a marked decline in GSH activity, which was assessed using Elmann's method. A significant reduction in the GSH level in E. coli cells was observed following treatment with analogues 16b and 21e, highlighting their ability to deplete GSH in bacterial systems. A positive correlation was observed between the analogue concentration and depletion of GSH levels. At 8× MIC, 16b and 21e caused 47% and 39% reduction in GSH activity, respectively, signifying gradual breakdown of the bacterial oxidative defense system in response to these active analogues (Fig. 7c).
2.8.4. Lipid peroxidation.
Malondialdehyde (MDA), formed through the oxidation of unsaturated fatty acids by ROS, serves as a pivotal indicator to estimate lipid peroxidation and a biomarker for assessing oxidative stress in bacterial cells.62 Absorbance at 535 nm, in direct relation to MDA levels, served as a key parameter to assess the extent of lipid peroxidation.63 The production of MDA in E. coli was significantly enhanced in the presence of both analogues. Furthermore, MDA production increased progressively with rising concentrations of the analogues, reaching 62.5% increase for 16b and 53.1% for 21e at 8× MIC (Fig. 7d). Thus, both analogues effectively amplified MDA production, correlating with the escalation of ROS, which in turn expedited bacterial cell death.
2.9. Interactions with human serum albumin (HSA)
Human serum albumin (HSA), the most abundant plasma carrier protein, functions as both a transporter and a reservoir for substances in the bloodstream. Its conformational flexibility and multiple binding sites allow it to bind a wide range of endogenous molecules and pharmaceutical agents. These drugs interact within the hydrophobic cavity of HSA through hydrogen bonding, electrostatic forces, hydrophobic interactions, and covalent bonding.64 These binding interactions are instrumental in shaping the biological potency of drugs. Owing to its remarkable attributes, HSA is recognized as a crucial target in design of effective antibacterial agents.65 Therefore, spectroscopic methods have been utilized to explore binding affinities of analogues 16b and 21e towards HSA.
2.9.1. UV-visible studies.
HSA (10 µM) displayed a characteristic weak absorption band at λmax 280 nm, attributed to aromatic amino acids (Trp, Tyr, and Phe), in a phosphate buffer medium (pH 7.4, 298 K).66 Incremental addition of analogues 16b (0–11 µM) and 21e (0–12 µM) to HSA led to enhancement of the absorption band at 280 nm, with new bands emerging at 330 nm for 16b and at 315 nm for 21e, indicating an altered microenvironment around aromatic residues due to complexation (Fig. 8a and b). The absence of shifts in HSA's absorption maxima upon interaction with analogues implies a non-covalent mode of interaction. This may be due to the π–π stacking interactions between the aromatic rings of analogues and HSA binding site. Employing the Benesi–Hildebrand equation,67 the binding constants of 16b and 21e with HSA were determined as 1.20 × 104 M−1 and 1.39 × 104 M−1, respectively (eqn (1) and Fig. S69). The binding constants highlight the significant interaction of 16b and 21e with HSA, with the latter exhibiting a slightly greater affinity than the former. For effective drug delivery, binding should be neither too strong nor too weak—excessively strong binding may hinder drug release at the target site, while overly weak binding could lead to premature dissociation, preventing the drug from reaching its intended destination. Therefore, these binding constants indicate an optimal balance between drug binding and release, ensuring effective delivery to the target site.| | 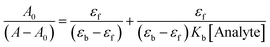 | (1) |
A0 represents the absorbance value of analogues 16b and 21e/HSA in a free state, while A signifies the absorbance value in the fully bound form of HSA with analogues 16b and 21e. εf and εb denote the molar extinction coefficients of analogues 16b and 21e/HSA in the absence and presence of the analyte, respectively. The binding constant (Kb) values were derived from the ratio of the intercept to the slope of the plot [A0/(A − A0) vs. 1/Analyte].
 |
| | Fig. 8 Spectral profiles of HSA, showing absorption and emission patterns with increasing concentrations of (a) and (c) 16b and (b) and (d) 21e in phosphate buffer (pH 7.4) at 298 K. | |
2.9.2. Fluorescence studies.
Fluorescence quenching studies provide meaningful insights into structural changes in proteins upon drug binding. HSA emits strong endogenous fluorescence with excitation at 280 nm and emission at 340 nm, owing to energy transfer among its dominant fluorescent groups including phenylalanine, tyrosine, and tryptophan. Among these, the fluorescence intensities of phenylalanine and tyrosine are often overlooked due to their minimal contribution. In particular, tryptophan (Trp 214), positioned in the hydrophobic region of subdomain IIA, serves as the main source of HSA's fluorescence.68 Hence, to further elucidate the interaction mechanism, emission studies were undertaken to assess the binding behaviour of 16b and 21e with HSA. The fluorescence intensity of HSA (10 µM) was progressively quenched upon incremental addition of 16b (0–15 µM) and 21e (0–36 µM) (Fig. 8c and d) at 298 K, potentially due to alterations in its microenvironment caused by foreign ligand interactions. Notably, upon incremental addition of 16b, a spectral shift was observed, marked with a new band at 460 nm and an isosbestic point at 415 nm. Additionally, to substantiate the quenching mechanism, temperature-dependent Stern–Volmer analyses were also conducted at 308 and 318 K (Fig. S70 and S71). To elucidate whether the quenching mechanism was dynamic or static, the Stern–Volmer equation (eqn (2)) was employed, to enable the analysis of quenching constants (KSV) and bimolecular quenching rates (Kq) (Fig. S72, S73 and Table 4). The calculated values of KSV and Kq at 298 K for 16b were 4.80 × 105 M−1 and 4.80 × 1013 M−1 s−1, respectively, while those for 21e were 4.89 × 105 M−1 and 4.89 × 1013 M−1 s−1, respectively. At high analogue concentrations, HSA's emission intensity was completely quenched, with substantial Ksv values confirming their strong quenching efficiency and binding affinity to HSA. Since the calculated Kq values exceeded the upper limit for dynamic quenching (2 × 1010 M−1 s−1), the results suggest a static quenching mechanism in both cases.69 Furthermore, elevated temperatures led to a decline in KSV values, further corroborating the static quenching behaviour of both analogues.
Table 4 Quenching and binding parameters for interactions of HSA with analogues 16b and 21e at various temperatures
| Analogues |
T (K) |
K
sv (× 105) (M−1) |
K
q (× 1013) (M−1 s−1) |
R
2
|
K
bin (× 106) M−1 |
n
|
R
2
|
|
16b
|
298 |
4.80 |
4.80 |
0.9729 |
6.06 |
1.55 |
0.9955 |
| 308 |
2.31 |
2.31 |
0.9833 |
3.33 |
1.25 |
0.9873 |
| 318 |
1.33 |
1.33 |
0.9817 |
1.98 |
1.18 |
0.9860 |
|
21e
|
298 |
4.89 |
4.89 |
0.9891 |
8.39 |
1.36 |
0.9964 |
| 308 |
4.02 |
4.02 |
0.9837 |
3.06 |
1.26 |
0.9924 |
| 318 |
3.45 |
3.45 |
0.9886 |
1.03 |
1.14 |
0.9961 |
In addition, a modified Stern–Volmer equation (eqn (3)) was applied to compute the binding constants (Kbin) at 298 K, which were determined to be 6.60 × 106 M−1 for 16b and 8.39 × 106 M−1 for 21e (Fig. S74, S75 and Table 4). These binding constants revealed a pronounced affinity between analogues and HSA, ensuring efficient drug delivery and timely release from serum albumin. Since n values slightly exceed unity across different temperatures, the findings indicate that the analogues primarily bind to a dominant high-affinity site, alongside the possibility of additional weaker interactions within HSA. A consistent drop in Kbin values with increasing temperature indicated a decline in HSA-analogue complex stability, eventually resulting in degradation. Typically, the interaction between small molecules and macromolecules is largely regulated by non-covalent forces, encompassing hydrogen bonding, van der Waals attractions, and hydrophobic and electrostatic interactions. To quantify these binding forces, the van't Hoff equation (eqn (4)) was applied to the interaction of analogues 16b and 21e with HSA. The entropy change (ΔS) and enthalpy change (ΔH) for both analogues were evaluated using the slope and intercept of the van’t Hoff plot (Table 5 and Fig. S76), while the binding free energy (ΔG) was calculated using eqn (5). The spontaneity of the interaction was evidenced by the negative ΔG value, with the corresponding ΔH and ΔS values also being negative, reinforcing the role of van der Waals forces and hydrogen bonding in the complexation of 16b and 21e with HSA.70
| |  | (2) |
| |  | (3) |
where
F0 and
F correspond to the fluorescence intensities of unbound and bound forms of HSA, respectively, with the analogues (
16b and
21e) serving as quenchers. The Stern–Volmer quenching constant value (
Ksv) was computed from the ratio of slope to intercept in
F0/
F and [analogues
16b and
21e] plots. To determine the bimolecular quenching rate constant (
Kq), the average value of the fluorescence lifetime (
τ0 = 10
−8 s) was employed. Furthermore, the binding constants (
Kb) and the number of binding sites (
n) were determined from the slope and intercept of the log [(
F0 −
F)/
F] and log [Analyte] plot.
Table 5 Thermodynamic parameters of binding of HSA with analogues 16b and 21e
| Analogues |
T (K) |
ΔH (kcal mol−1) |
ΔS (cal mol−1 K−1) |
ΔG (kcal mol−1) |
R
2
|
|
16b
|
298 |
−11.41 |
−7.05 |
−9.31 |
0.9974 |
| 308 |
−9.24 |
| 318 |
−9.17 |
|
21e
|
298 |
−19.74 |
−34.42 |
−9.48 |
0.9976 |
| 308 |
−9.14 |
| 318 |
−8.79 |
The thermodynamic parameters were determined based on the van't Hoff equations (eqn (4) and (5)).
| | 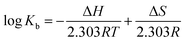 | (4) |
R denotes the gas constant, and
T signifies the temperature (Kelvin). A graph correlating log
Kb and 1/
T was plotted to evaluate the entropy change (Δ
S) and enthalpy change (Δ
H).
2.9.3. Time-correlated single photon counting (TCSPC) experiment.
Furthermore, a time-correlated single photon counting experiment was performed to elucidate the quenching mechanism governing the interaction between analogues and HSA. Steady-state emission studies indicated that the binding interactions between HSA and analogues were predominantly driven by the static quenching mechanism. To validate this, time-resolved spectra of native HSA (10 µM) were recorded both individually and with incremental addition of analogues 16b and 21e (10–40 µM). A marked decline in the time decay profile of native HSA was noted with the introduction of analogues 16b and 21e, suggesting the occurrence of dynamic quenching potentially resulting from collisional deactivation (Fig. 9a, b and Table S1).71 Drawing upon the integrated findings from both steady-state fluorescence and time-resolved studies, the interactions of analogues 16b and 21e with HSA appears to be governed by both static and dynamic quenching mechanisms. The average fluorescence lifetimes (τav) for triexponential iterative fittings were assessed using the described equation (eqn (6)):| |  | (6) |
where αi is the pre-exponential factor and τi is the decay time of the analogue in free and bound states. The chi-square (χ2) values were close to unity, suggesting a good fit.
 |
| | Fig. 9 Change in lifetime spectra of HSA (10 µM) with increasing concentrations (10–40 µM) of analogues (a) 16b and (b) 21e in phosphate buffer (pH 7.4). | |
In addition, refolding studies were conducted using the anionic surfactant sodium dodecyl sulfate (SDS), known for its ability to deactivate and denature proteins, to investigate the reversibility of dihydrofurocoumarin analogues binding to HSA.72 Fluorescence spectroscopic analysis indicated that active analogues 16b and 21e influenced the conformational dynamics of serum albumin. SDS facilitated a moderate restoration of HSA's native structure by displacing the active analogues bound to the serum albumin. Hence, the findings suggest that these analogues reversibly interact with HSA, thereby contributing to their favorable pharmacokinetic profile (Fig. S77).
2.9.4. Circular dichroism (CD) spectroscopy.
Circular dichroism (CD) offers a prompt and efficacious approach for investigating protein–ligand interactions, particularly for evaluating the secondary structural elements of proteins and detecting conformational changes induced by ligand binding. The CD spectrum of HSA reveals two distinct negative ellipticity bands—one at 208 nm, corresponding to the π → π* electronic transitions, and another at 222 nm, attributed to n → π* transitions of the peptide bonds (Fig. 10). These spectral transitions are characteristic of the α-helical structure of the human serum albumin.73
 |
| | Fig. 10 CD spectra of 2.5 µM HSA in its free form and in the presence of 0.31 µM 16b and 21e. | |
Mean residue ellipticity (MRE) and % α-helicity were calculated using standard equations (eqn (S8) and (S9), provided in the SI). The α-helical content of HSA was found to be 64.72%. Upon addition of 16b, the α-helical content of HSA increased to 81.87%, thereby influencing its secondary conformation.74 Conversely, 21e reduced the α-helical content to 26.16%, indicating a loss of helicity and possible protein destabilization.75 Collectively, these observations imply that both analogues are capable of inducing conformational changes in HSA (Table 6).
Table 6 α-Helical content (%) of HSA calculated from CD spectra
| Complex |
α-Helical content (%) |
| HSA (free) |
64.72 |
| HSA + 16b |
81.87 |
| HSA + 21e |
26.16 |
2.9.5. Drug displacement studies.
Drug displacement studies utilizing warfarin, ibuprofen, and bilirubin as site markers were conducted to precisely map the binding sites of analogues on HSA. The two-principal drug-binding domains of HSA are Sudlow site I, known for its affinity toward warfarin, and Sudlow site II, which binds ibuprofen. Site I, located in domain IIA, and Site II, found within domain IIIA, play key roles in mediating drug transport through the vascular system. Moreover, a distinct bilirubin specific binding site, positioned within subdomain IB, has recently been discovered, which also exhibits selective affinity toward hemin and fusidic acid.76 To explore the possible binding sites of analogues 16b and 21e, fluorescence titration assays were performed at an excitation wavelength of 280 nm, using HSA (10 µM) and respective site markers, with incremental addition of the analogues (Fig. S78 and S79). The emission band at 350 nm, observed for the HSA-site marker complexes with the analogues, was efficiently quenched in both cases. Notably, a distinct emission band characteristic of 16b emerged at 460 nm, accompanied by an isosbestic point at 415 nm. The binding constants (Kbin) were determined using the modified Stern–Volmer equation (Fig. S80, S81 and Table S2). Notably, the Kbin of HSA decreased significantly in the presence of warfarin for 16b and bilirubin for 21e, suggesting that pre-occupancy of these sites diminishes the analogues' binding affinity to HSA. This competitive behavior highlights striking site selectivity, with 16b exhibiting a dominant affinity for Sudlow site I, while 21e preferentially engages with the heme site on HSA.
2.9.6. Molecular docking.
Molecular docking has ascended as an indispensable facet for in silico computational drug design, offering unparalleled insights into ligand-receptor dynamics.77 So, the molecular docking study was conducted to validate the drug displacement results, identify the potential binding sites of analogues 16b and 21e within HSA using its crystal structure (PDB ID: 1N5U) as the designated target and explore the interactive forces governing their binding to serum albumin. The ligand–HSA interactions were set up via the AutoDock Tools graphical user interface (1.5.6rc3), involving the elimination of water molecules, the addition of polar hydrogen atoms, and the determination of Gasteiger charges. Furthermore, visualization analyses were conducted using the Discovery Studio.
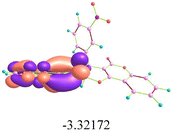
Docking simulations of HSA with analogues 16b and 21e revealed the best binding conformations, with minimum binding energy values of −7.26 kcal mol−1 and −10.36 kcal mol−1, respectively (Table S3). Docking results indicated that 16b was confined within the hydrophobic cavity of subdomain IIA (warfarin binding site) of HSA, encased by key amino acid residues, including TYR A:148, SER A:193, GLN A:196, ARG A:197, CYS A:200, and CYS A:246 (Fig. 11). The naphthalimide moiety of 16b engaged in notable π–π stacking interactions with TYR A:148 of HSA, while CYS A:246 mediated π-alkyl interactions with the coumarin scaffold. GLN A:196 and SER A:193 residues of HSA interacted with fluorophenyl, forming halogen bonds with bond distances of 3.07 Å and 3.43 Å, respectively. Moreover, CYS A:200 participated in π–sulfur interactions, further reinforcing the stability of the complex. In comparison, 21e was trapped within the heme site located in sub-domain IB (bilirubin binding site) of HSA, where it interacted with key amino acid residues, including LEU A:115, VAL A:116, ARG A:117, TYR A:138, ARG A:186, and LYS A:190 (Fig. 12). Analysis of the binding pattern revealed that the oxygen atom of the nitro group (d = 2.01 Å) and the oxygen atom of the carbonyl group in the coumarin moiety (d = 2.03 Å) formed conventional hydrogen bonds with LYS A:190, while TYR A:138 engaged in π–π stacking interactions with the coumarin ring (d = 4.07 Å, 5.63 Å). Beyond these interactions, the complex's stability is further reinforced by π–cation and π–alkyl interactions. Therefore, these diverse non-covalent forces collectively contribute to enhanced stability of the HSA–16b/21e complex, highlighting its strong potential for binding with the HSA protein.
 |
| | Fig. 11 (a) Analogue 16b docked in the binding pocket of HSA; (b) 2D representation of 16b–HSA interaction; (c) 3D depiction of optimum 16b–HSA conformation; (d) 16b confined within the hydrophobic cavity of HSA. | |
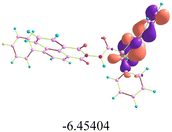
 |
| | Fig. 12 (a) Analogue 21e docked in the binding pocket of HSA; (b) 2D representation of 21e–HSA interaction; (c) 3D depiction of optimum 21e–HSA conformation; (d) 21e confined within the hydrophobic cavity of HSA. | |
Additionally, site-specific docking was performed using sequences designated for subdomains IIA, IIIA, and IB to achieve more precise determination of binding sites (Table S4 and Fig. S82, S83). Among these sites, 16b docked at subdomain IIA of HSA exhibited the lowest binding energy (−8.86 kcal mol−1), compared to −6.65 kcal mol−1 at subdomain IIIA and −8.35 kcal mol−1 at subdomain IB. In comparison, 21e docked at subdomain IB of HSA, where it exhibited the lowest binding energy (−10.80 kcal mol−1) compared to −8.88 kcal mol−1 at subdomain IIA and −8.96 kcal mol−1 at subdomain IIIA (Table 7). In-depth analysis of free binding energies of 16b and 21e across all three sites provides valuable insight into their binding preferences, revealing that 16b preferentially binds to Suldow site I, located within the hydrophobic cavity of subdomain IIA, whereas 21e favors the heme site within subdomain IB. Thus, the molecular docking results of serum albumin crystal structures with analogues 16b and 21e exhibit strong concordance with the site marker drug displacement experiment, validating their specific binding behavior.
Table 7 Comparative docking analysis of analogues 16b and 21e at subdomains IIA, IIIA, and IB of HSA
| Binding sites |
Reference ligand |
Sub-domain |
Amino acid residues |
Affinity (kcal mol−1) |
|
16b
|
21e
|
| Sudlow site I |
Warfarin |
IIA |
197–297 |
−8.86 |
−8.88 |
| Sudlow site II |
Ibuprofen |
IIIA |
384–497 |
−6.65 |
−8.96 |
| Heme site |
Bilirubin |
IB |
108–196 |
−8.35 |
−10.80 |
2.10. DNA binding studies
Bacterial DNA serves as a vital biomacromolecule, essential for bacterial growth and survival, and has been widely recognized as a highly promising target for drug intervention. The interaction of drug molecules with bacterial DNA frequently results in different extents of DNA damage.78 Since bacterial cell death was triggered by analogues 16b and 21e through mechanisms involving membrane disruption and oxidative stress, their potential interaction with DNA was probed using spectroscopic techniques and substantiated by molecular docking studies.
Absorption spectroscopy was utilized to monitor hyperchromic and hypochromic effects in DNA, which serve as characteristic indicators for alterations in the double-helical structure. The absorption spectra of the active analogues were recorded in PBS (pH 7.4) with successive additions of ct-DNA. For both analogues 16b and 21e, incremental addition of ct-DNA (0–30 µM) resulted in a hyperchromic shift at 260 nm along with hypochromic shifts at 360 nm and 310 nm, respectively (Fig. 13). The binding constants (Kb) for DNA interaction, determined using the Benesi–Hildebrand equation, were 1.3 × 104 M−1 and 0.8 × 104 M−1 for 16b and 21e, respectively, indicating binding of the analogues to ct-DNA (Fig. S84). Moreover, to better understand the binding mode of the analogues with ct-DNA, particularly to differentiate between intercalation and groove binding, an ethidium bromide (EB) displacement experiment was performed. Incremental addition of the active analogues to the EB–ct-DNA complex (3 µM:30 µM) led to gradual decrease in fluorescence intensity at 602 nm, indicating EB displacement from DNA and providing evidence for an intercalative binding mode that can impede DNA replication (Fig. S85).
 |
| | Fig. 13 Absorption spectra of (a) 16b and (b) 21e in phosphate buffer (pH 7.4) at 298 K upon progressive addition of ct-DNA (0–30 µM). | |
Furthermore, the active analogues were docked with ct-DNA (PDB: 1BNA) using AutoDock, and the resulting interactions were interpreted using Discovery Studio. The docking results demonstrated minimum binding energies of −11.18 kcal mol−1 for 16b and −10.22 kcal mol−1 for 21e (Table S5). Fig. 14a and b illustrates that the naphthalimide ring interacts with DC21 (Chain B, d = 5.61 and 3.48 Å) through π–sigma and π–π T-shaped interactions. The amide linker establishes conventional hydrogen bonding with DG22 (Chain B, d = 1.77 and 3.64 Å). The carbonyl oxygen of the naphthalimide engages with DA5 (Chain A, d = 2.02 Å) and DG4 (Chain A, d = 2.90 Å), while the oxygen of the dihydrofurocoumarin moiety also forms a conventional hydrogen bond with DG4 (Chain A, d = 1.92 Å). On the other hand, in 21e, the oxygen atom of the nitro group forms a carbon–hydrogen bond with DC15 (Chain B, d = 3.27 Å). The oxygen and carbonyl groups of the coumarin scaffold establish conventional hydrogen bonds with DG10 (Chain A, d = 2.12 Å) and DG11 (Chain A, d = 1.62 Å), respectively. Additionally, the coumarin ring participates in π–π T-shaped stacking with DA17 (Chain B, d = 5.87 Å) and forms a π-donor hydrogen bond with DG16 (Chain B, d = 2.62 Å), which further stabilizes the complex. An unfavorable acceptor–acceptor interaction was also observed between the carbonyl oxygen of the ligand and DC11 (Chain A, d = 2.31 Å), which could influence the overall binding stability (Fig. 14c and d). Thus, the favorable docking score, together with key molecular interactions, underscores the enhanced stability of the complexes of active analogues and highlights their potential in bacterial growth inhibition.
 |
| | Fig. 14 3D and 2D docked poses of 16b (a) and (b) and 21e (c) and (d) with DNA (PDB: 1BNA). | |
2.11. Cytotoxicity assay
Cytotoxicity assessment in normal mammalian cell lines is a crucial prerequisite for ensuring the safety of novel drug candidates, as low or non-toxicity is vital for their clinical applicability.79 Consequently, the colorimetric MTT assay was employed to analyze the cell viability of both active analogues against HEK293 (human kidney) cells over a concentration range of 0.01 to 100 µM. Cell viability assessments revealed that 16b exhibited values of 94.3, 91.2, 88.7, 85.4 and 81.7%, whereas 21e showed viability levels of 96.8, 93.2, 91.6, 89.7 and 86.4%, corresponding to concentrations of 0.01, 0.1, 1, 10, and 100 µM, respectively (Fig. 15). Analogue 16b retained a maximum cell viability of 81.7%, while 21e exhibited 86.4% toward mammalian HEK293 cell lines, even at a higher concentration of 100 µM. The low cytotoxicity values indicate excellent selectivity in targeting bacterial cells over normal cells, highlighting their biosafety and affirming their suitability for further investigation as antibacterial agents.
 |
| | Fig. 15 % Cell viability of analogues 16b and 21e against the normal cell line HEK293. *p < 0.05; **p < 0.01; ***p < 0.001; n = 3. | |
2.12. Quantum chemical studies
The HOMO–LUMO energy gap (ΔE) is a crucial quantum descriptor, often employed to assess the chemical reactivity and stability of compounds. In the context of antimicrobial research, favorable ΔE values may aid in identifying promising candidates with enhanced bioactive potential.80 To further explore the electronic characteristics underlying this potential, the frontier molecular orbital distributions of analogues 16b and 21e were examined. For both analogues, the HOMO was predominantly localized around the dihydrofurocoumarin ring, suggesting its interaction with positively charged biological ions through electrostatic forces. In contrast, the LUMO was positioned on the naphthalimide ring in 16b and on the coumarin ring in 21e, indicating their potential interactions with negatively charged residues. A smaller HOMO–LUMO energy gap observed for both analogues may facilitate more efficient electronic excitation between donor and acceptor orbitals, potentially enhancing molecular reactivity and interactions with biomolecules. Moreover, computational results further indicated that both analogues fell outside the phototoxic window (ΔE = 7.2 ± 0.4 eV), suggesting that they are unlikely to induce phototoxicity.
Molecular electrostatic potential (MEP) maps offer qualitative insights into chemical reactivity and selectivity by illustrating the spatial distribution of electrostatic potential, which reflects a molecule's electronic structure and shape.81 Analysis of the MEP surfaces revealed that the regions highlighted in red corresponding to electron-rich areas were primarily localized on the oxygen atoms of 16b and 21e, which could serve as potential sites for interactions with biological targets (Table 8 and Fig. S6).
Table 8 Atomic orbital HOMO–LUMO compositions, related energy gaps, and MEP of analogues 16b and 21e
| Analogues |
16b
|
21e
|
| LUMO (eV) |
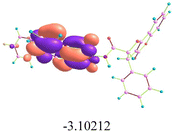
|
|
| HOMO (eV) |
|

|
| MEPs |

|

|
| ΔE (energy gaps in eV) |
3. Conclusion
In summary, a series of dihydrofurocoumarin analogues were synthesized through a multistep synthetic route, culminating in multi-component cascade reaction, as potential antibacterial agents to counteract the escalating threat of drug resistance. A comprehensive evaluation against eight Gram-positive and Gram-negative bacterial strains was conducted to assess the antimicrobial potential of the synthesized analogues. Preliminary bioactivity and structure–activity relationship analyses revealed that certain derivatives exhibited profound bacterial growth inhibition. Remarkably, analogues 16b and 21e, functionalized with electron-withdrawing groups, displayed superior activity, with an MIC value of 1.56 µg mL−1, surpassing that of clinical amoxicillin against E. coli. Both active analogues 16b and 21e, exhibiting low cytotoxicity, effectively impede biofilm formation, consequently delaying the onset of drug resistance. Furthermore, biofunctional evaluation reveals that these analogues induce intracellular ROS generation, leading to oxidative stress accumulation and the oxidation of GSH to GSSG, thereby impairing GSH activity and causing cellular oxidative damage. HSA studies demonstrated that both analogues are adeptly transported to the target site via carrier proteins. Site marker drug displacement experiments affirmed the preferential binding of 16b to Sudlow site I and 21e to the heme site on HSA. These findings were further reinforced by molecular docking studies, which showed strong congruence with experimental data. In addition, the active analogues intercalate into DNA, forming a supramolecular complex that inhibits bacterial replication. Moreover, in silico ADME analysis confirmed that both analogues possess favorable pharmacokinetic profiles, while quantum chemical studies further supported the superior bioactivity of naphthalimide containing 16b over 21e, as evidenced by its narrower FMO energy gap. The multitarget mechanisms identified in these analogues acted synergistically to enhance their antibacterial potency. These promising insights pave the way for further refinement and clinical exploration of dihydrofurocoumarins as effective broad-spectrum antibacterial candidates.
4. Experimental section
4.1. Chemistry
The chemicals and solvents used in both chemical and biological experiments were obtained commercially from Sigma Aldrich, Spectrochem, and Loba-Chemie and were used without any additional purification. Melting points were noted using EQ 730A melting point apparatus with open capillaries and were uncorrected. Thin-layer chromatography (TLC) analysis was performed using precoated silica gel HF-254 plates and the analogues were purified by column chromatography on silica gel (60–120/100–200 mesh) using chloroform and ethyl acetate as eluents. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance Neo 500 MHz spectrometer, with TMS as the internal standard. Chemical shifts (δ) were reported in parts per million (ppm), coupling constants (J) were expressed in hertz (Hz), and signals were characterized as singlets (s), doublets (d), triplets (t), and multiplets (m). The high-resolution mass spectra of the analogues were obtained with a Waters Xevo G2-XS QTof mass spectrometer featuring an electrospray ionization source confirming the analogues' purity to be greater than 95%. Bacterial studies were carried out using a Biotek ELISA plate. Referred UV and fluorescence spectra were recorded using Shimadzu UV visible and Varian Cary Eclipse fluorescence spectrophotometers, respectively. All fluorescence micrographs were obtained using a Confocal Laser Scanning Microscope (Olympus FV1200). CD spectra were recorded on a JASCO J-1500 spectrometer. Scanning electron microscopy (SEM) was conducted using an SU 8010 Series (Hitachi). Sample masses were measured on a microbalance with a precision of 0.1 mg. Stock solutions of analogues 16b and 21e were prepared in DMSO at 800 µg mL−1. Serial two-fold dilutions were then made to obtain the following concentrations for MIC determination: 800, 400, 200, 100, 50, 25, 12.5, 6.25, 3.12, and 1.56 µg mL−1. DMSO was used as a negative control in all experiments. For HSA and DNA binding studies, stock solutions of 16b and 21e were prepared in DMSO at 10−3 M.
4.2. General procedure for the synthesis of intermediates
4.2.1. Synthesis of benzo[de]isochromene-1,3-dione (2)82.
Under continuous stirring in an ice bath, potassium dichromate (10 g, 65 mmol) was gradually added to a solution of acenaphthene 1 (2.0 g, 12.98 mmol) in 50 mL of acetic acid. The reaction mixture was first stirred at room temperature for 25 min and then refluxed for 3 h. Upon completion of the reaction, as confirmed by TLC, 250 mL of water was added, resulting in precipitation of white solid 2 in 95% yield; mp 264–266 °C.
4.2.2. Synthesis of 2-amino-1H-benzo[de]isoquinoline1,3(2H)-dione (3)82.
A vigorously stirred solution of analogue 2 (1 g, 5.05 mmol) in 30 mL of ethanol was treated with hydrazine hydrate (506 mg, 10.1 mmol) and allowed to stir at room temperature for 30 min. Upon completion of the reaction, the addition of water induced precipitation. The resulting solid was filtered, thoroughly washed with water to remove residual hydrazine hydrate, and dried to afford a yellow solid product 3 in 84% yield; mp 270–272 °C.
4.2.3. Synthesis of 2-bromo-N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (4).
Bromoacetyl bromide (807 mg, 4 mmol) was added to a stirred solution of analogue 3 (1 g, 4.7 mmol) in 1,2-dichloroethane (10 mL), and the resulting mixture was refluxed for 10 min. Upon completion of the reaction, the reaction mixture was poured into cold water and precipitates were filtered and washed with cold MeOH to yield pure 4. White solid; 89% yield; mp 235–237 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.24 (s, 1H), 8.66 (dd, J = 7.4, 0.5 Hz, 2H), 8.35 (d, J = 7.9 Hz, 2H), 7.83 (t, J = 7.8 Hz, 2H), 4.20 (s, 2H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 168.2, 163.2, 136.7, 133.7, 131.9, 128.2, 127.6, 120.9, 25.3.
4.3. General procedure for naphthalimide tethered dihydrofurocoumarin analogue (7a–f)
A mixture of 4 (500 mg, 1.5 mmol), coumarin 5 (244 mg, 1.5 mmol) and various aldehydes 6a–f (1.5 mmol) in pyridine (5 mL) was stirred at 80 °C for 5–6 h. After completion of the reaction, cold water was added to the reaction mixture, and the residue obtained was filtered, vacuum-dried, and triturated followed by washing with MeOH to obtain the pure analogue 7a–f.
4.3.1. Spectral data of N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (7a).
White solid; 83% yield; mp 247–249 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.46 (s, 1H), 8.58 (d, J = 7.2 Hz, 1H), 8.55 (d, J = 7.2 Hz, 1H), 8.50 (d, J = 8.2 Hz, 2H), 7.94 (d, J = 7.7 Hz, 1H), 7.92–7.88 (m, 2H), 7.75 (t, J = 7.3 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.43–7.39 (m, 4H), 7.36–7.33 (m, 1H), 5.71 (d, J = 5.7 Hz, 1H), 4.88 (d, J = 5.7 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.5, 166.2, 161.4, 158.3, 154.7, 139.6, 135.4, 133.4, 131.7, 131.6, 131.4, 128.9, 127.7, 127.6, 127.4, 127.1, 124.4, 123.5, 121.4, 121.3, 116.6, 111.6, 104.3, 89.1, 50.7; HRMS (ESI): calcd for C30H18N2O6 [M + H]+, 503.1198, found, 503.1240.
4.3.2. Spectral data of N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-(p-tolyl)-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (7b).
Off white solid; 81% yield; mp 270–272 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.45 (s, 1H), 8.59 (dd, J = 18.7, 7.2 Hz, 2H), 8.54 (d, J = 8.2 Hz, 2H), 7.96–7.91 (m, 3H), 7.77 (t, J = 7.5 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.28 (d, J = 7.9 Hz, 2H), 7.23 (d, J = 7.8 Hz, 2H), 5.66 (d, J = 5.7 Hz, 1H), 4.84 (d, J = 5.6 Hz, 1H), 2.32 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.5, 166.1, 161.4, 158.4, 154.7, 137.0, 136.7, 135.4, 133.4, 131.8, 131.7, 131.5, 129.4, 127.5, 127.2, 124.5, 123.5, 121.5, 121.4, 116.6, 111.7, 104.4, 89.2, 50.4, 20.6; HRMS (ESI): calcd for C31H20N2O6 [M + H]+, 517.1355, found, 517.1393.
4.3.3. Spectral data of 3-(4-chlorophenyl)-N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (7c).
White solid; 76% yield; mp 260–263 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.44 (s, 1H), 8.61 (d, J = 7.3 Hz, 1H), 8.56 (dd, J = 10.8, 8.0 Hz, 3H), 7.96–7.91 (m, 3H), 7.78 (t, J = 7.8 Hz, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.48 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 5.72 (d, J = 5.9 Hz, 1H), 4.87 (d, J = 5.9 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.4, 166.4, 161.4, 158.3, 154.7, 138.4, 135.4, 133.5, 132.4, 131.8, 131.7, 131.5, 129.6, 128.8, 127.5, 127.2, 124.5, 123.6, 121.4, 121.3, 116.7, 111.6, 103.9, 88.8, 50.1; HRMS (ESI): calcd for C30H17ClN2O6 [M + H]+, 537.0809, found, 537.0850.
4.3.4. Spectral data of N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-3-(4-methoxyphenyl)-4-oxo-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (7d).
Off white solid; 88% yield; mp 253–255 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 8.62–8.53 (m, 4H), 7.96–7.91 (m, 3H), 7.78–7.74 (m, 1H), 7.54 (dd, J = 8.4, 3.6 Hz, 1H), 7.50–7.47 (m, 1H), 7.30 (d, J = 8.7 Hz, 2H), 6.96 (d, J = 8.6 Hz, 2H), 5.63 (d, J = 5.7 Hz, 1H), 4.80 (dd, J = 5.7, 2.1 Hz, 1H), 3.76 (d, J = 2.1 Hz, 3H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.5, 165.9, 161.4, 158.7, 158.3, 154.6, 135.3, 133.3, 131.7, 131.6, 131.5, 128.6, 127.4, 127.3, 127.1, 125.7, 124.8, 124.4, 123.5, 121.4, 116.6, 114.4, 114.2, 114.0, 111.7, 104.4, 89.2, 55.1, 50.0; HRMS (ESI): calcd for C31H20N2O7 [M + H]+, 533.1304, found, 533.1344.
4.3.5. Spectral data of 3-(2,4-dichlorophenyl)-N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (7e).
White solid; 65% yield; mp 261–263 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 8.61 (d, J = 7.3 Hz, 1H), 8.58–8.55 (m, 3H), 7.96–7.92 (m, 3H), 7.79 (dd, J = 11.5, 4.3 Hz, 1H), 7.66 (dd, J = 7.1, 2.3 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.42–7.37 (m, 2H), 5.76 (d, J = 5.1 Hz, 1H), 5.37 (d, J = 4.9 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 166.8, 161.4, 161.3, 160.3, 158.2, 154.8, 135.3, 134.7, 133.7, 132.2, 131.7, 131.6, 131.5, 131.2, 131.0, 130.0, 128.9, 128.7, 127.5, 127.3, 127.2, 124.6, 123.5, 122.3, 121.6, 121.5, 116.8, 111.5; HRMS (ESI): calcd for C30H16Cl2N2O6 [M + H]+, 571.0385, found, 571.0461.
4.3.6. Spectral data of 3-(4-bromophenyl)-N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (7f).
White solid; 73% yield; mp 285–288 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.43 (s, 1H), 8.61 (dd, J = 7.3, 1.0 Hz, 1H), 8.57 (dd, J = 11.7, 4.6 Hz, 3H), 7.96–7.92 (m, 3H), 7.79–7.76 (m, 1H), 7.62–7.59 (m, 2H), 7.55 (d, J = 8.1 Hz, 1H), 7.52–7.48 (m, 1H), 7.37 (d, J = 8.5 Hz, 2H), 5.71 (d, J = 6.0 Hz, 1H), 4.84 (d, J = 6.0 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.3, 166.4, 161.4, 158.3, 154.7, 138.9, 135.4, 133.5, 131.8, 131.7, 131.5, 129.9, 127.5, 127.2, 124.5, 123.6, 121.4, 121.3, 120.9, 116.7, 111.6, 103.8, 88.7, 50.2; HRMS (ESI): calcd for C30H17BrN2O6 [M + H]+, 581.0270, found, 581.0350.
4.4. General procedure for synthesis of intermediates
4.4.1. Synthesis of 5-bromo-1,2-dihydroacenaphthylene (8).
Acenaphthene 1 (10 g, 64.8 mmol) was dissolved in 15 mL of DMF and stirred at room temperature, followed by the gradual addition of N-bromosuccinimide (11.53 g, 64.8 mmol) in portions. The mixture was continuously stirred for 3 h, with reaction progress tracked via TLC. Upon completion of the reaction, 100 mL of cold water was added, prompting the formation of yellow precipitates. The solution was filtered, thoroughly washed with water and vacuum-dried to obtain yellow product 8 in 95% yield; mp 53–56 °C.
4.4.2. Synthesis of 6-bromo-1H,3H-benzo[de]isochromene-1,3-dione (9).
Sodium dichromate dihydrate (12.5 g, 41.94 mmol) was gradually added to a cooled solution of analogue 8 (2 g, 8.6 mmol) in 50 mL of glacial acetic acid at 0 °C. The reaction mixture was stirred at room temperature for 10 min followed by refluxing for 4 h. After cooling, the mixture was poured into 500 mL of ice-cold water, leading to precipitation of the green solid. The precipitate was washed with hot water to remove any remaining chromium salts, affording a white solid product 9 in 82% yield; mp 222–225 °C.
4.4.3. Synthesis of 2-amino-6-bromo-1H-benzo[de]isoquinoline-1,3(2H)-dione (10).
Hydrazine hydrate (361 mg, 7.22 mmol) was introduced into a stirred ethanolic solution (20 mL) of 9 (1 g, 3.61 mmol). The reaction was stirred at room temperature for 30 min. Upon completion of the reaction, water was added, causing precipitation. The resulting solid was filtered, washed extensively with water to remove excess hydrazine hydrate, and dried to obtain yellow product 10 in 84% yield; mp 270–272 °C.
4.4.4. Synthesis of 2-amino-6-morpholino-1H-benzo[de]isoquinoline-1,3(2H)-dione (11).
To a solution of analogue 10 (1 g, 3.45 mmol) in 10 mL of DMF, morpholine (888.62 mg, 10.2 mmol) and potassium carbonate (553 mg, 4 mmol) were added, and the reaction mixture was heated at 100 °C for 3 h. Once cooled, 100 mL of water was introduced, leading to precipitation. The crude product was separated via filtration, washed with cold MeOH, and directly employed in the next step.
4.4.5. Synthesis of 2-bromo-N-(6-morpholino-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (12).
To a stirred solution of crude analogue 11 (1 g, 3.36 mmol) in 10 mL of 1,2-dichloroethane, bromoacetyl bromide (574 mg, 2.85 mmol) was added, and the reaction mixture was refluxed for 10 min. After completion of the reaction, the mixture was poured into cold water, causing precipitation. The solid was filtered, washed thoroughly with cold methanol, and dried to yield pure solid product 12. Mustard solid; 85% yield; mp 228–230 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.42 (s, 1H), 8.75 (dd, J = 7.3, 0.6 Hz, 1H), 8.71 (d, J = 8.2 Hz, 1H), 8.67–8.64 (m, 1H), 7.91 (dd, J = 8.4, 7.5 Hz, 1H), 7.57 (d, J = 8.3 Hz, 1H), 4.31 (s, 4H), 4.20 (s, 2H), 3.64 (s, 4H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 169.5, 163.7, 163.2, 154.2, 135.4, 134.8, 132.0, 130.4, 128.0, 125.9, 121.9, 117.4, 117.3, 77.4, 77.1, 66.6, 54.3, 25.1.
4.5. General procedure for synthesis of a 6-amino-substituted naphthalimide tethered dihydrofurocoumarin analogue (13)
A reaction mixture containing 12 (1 g, 2.4 mmol), coumarin 5 (389 mg, 2.4 mmol) and benzaldehyde 6a (255 mg, 2.4 mmol) in pyridine (5 mL) was stirred at 80 °C for 5 h. After completion of the reaction, cold water was added to the reaction mixture, and the residue obtained was filtered, titrurated well with methanol and vacuum-dried. The crude product was purified using column chromatography (CHCl3:EtOAc 90:10) to yield the pure analogue N-(6-morpholino-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (13). Yellow solid; 69% yield; mp 275–278 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.36 (s, 1H), 8.58–8.47 (m, 3H), 7.95 (dd, J = 7.9, 1.5 Hz, 1H), 7.88–7.83 (m, 1H), 7.79–7.75 (m, 1H), 7.54 (d, J = 8.3 Hz, 1H), 7.49 (dd, J = 11.3, 4.0 Hz, 1H), 7.43–7.39 (m, 4H), 7.39–7.38 (m, 1H), 7.37–7.33 (m, 1H), 5.68 (d, J = 5.7 Hz, 1H), 4.86 (d, J = 5.7 Hz, 1H), 3.93–3.90 (m, 4H), 3.26 (s, 4H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.4, 166.2, 161.6, 161.0, 158.3, 156.3, 154.7, 139.7, 133.4, 133.2, 133.1, 131.8, 131.7, 131.6, 129.1, 128.9, 127.7, 127.6, 126.2, 125.3, 124.4, 123.6, 122.0, 121.9, 116.6, 115.2, 114.8, 111.7, 104.3, 89.1, 66.1, 53.0, 50.7; HRMS (ESI): calcd for C34H25N3O7 [M + H]+, 588.1726, found, 588.1772.
4.6. General procedure for the synthesis of intermediates
4.6.1. Synthesis of 6-aryl-substituted 2-amino-1H-benzo[de]isoquinoline-1,3(2H)-dione (14a–f)81.
Under an inert atmosphere, 10 (1 g, 3.45 mmol) and boronic acids (4.104 mmol) were dissolved in an acetonitrile/water (9:1) mixture. Potassium carbonate (708 mg, 5.12 mmol) and tetrakis(triphenylphosphine)palladium(0) (5 mol%) were introduced, and the reaction was refluxed for 3–4 h. After completion of the reaction, the crude product was separated, rinsed with warm water, dried, followed by washing with cold MeOH, and then directly used in the subsequent steps.
4.6.2. Synthesis of 6-aryl substituted 2-bromo-N-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (15a–f).
A reaction mixture of 14a–f (3.36 mmol) and bromoacetyl bromide (573 mg, 2.85 mmol) in 10 mL of 1,2-dichloroethane was refluxed for 10 min. After the reaction was completed, cold water was added, resulting in precipitation. The obtained solid was filtered, thoroughly washed with cold MeOH, and dried to yield pure product 15a–f.
4.6.2.1. Spectral data of 2-bromo-N-(1,3-dioxo-6-phenyl-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (15a).
Beige solid; 88% yield; mp 225–227 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.48 (s, 1H), 8.78–8.75 (m, 2H), 8.49 (dd, J = 8.5, 0.9 Hz, 1H), 7.85–7.81 (m, 2H), 7.59 (t, J = 6.2 Hz, 3H), 7.53–7.50 (m, 2H), 4.23 (s, 2H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 169.0, 163.7, 163.6, 150.7, 137.8, 136.1, 134.0, 133.6, 130.5, 129.7, 129.2, 128.9, 128.7, 128.5, 127.4, 120.5, 119.2, 24.6.
4.6.2.2. Spectral data of 2-bromo-N-(6-(4-fluorophenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (15b).
Off white solid; 85% yield; mp 237–238 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.46 (s, 1H), 8.78 (d, J = 7.6 Hz, 2H), 8.45 (d, J = 8.3 Hz, 1H), 7.84 (dd, J = 9.7, 7.9 Hz, 2H), 7.53–7.48 (m, 2H), 7.30 (t, J = 8.6 Hz, 2H), 4.23 (s, 2H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 169.0, 164.4, 163.7, 163.5, 149.4, 135.7, 134.0, 133.5, 133.3, 131.6, 131.5, 130.5, 128.7, 128.6, 127.6, 120.6, 119.4, 116.1, 115.9, 24.6.
4.6.2.3. Spectral data of 2-bromo-N-(6-(4-methoxyphenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (15c).
Light green solid; 88% yield; mp 240–243 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.48 (s, 1H), 8.78–8.74 (m, 2H), 8.53 (d, J = 8.5 Hz, 1H), 7.86–7.81 (m, 2H), 7.50 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.1 Hz, 2H), 4.23 (s, 2H), 4.03 (s, 3H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 169.1, 163.8, 163.6, 159.5, 150.2, 136.1, 134.0, 133.7, 131.3, 131.2, 130.5, 128.8, 128.5, 127.4, 120.5, 118.9, 114.7, 55.8, 24.6.
4.6.2.4. Spectral data of 2-bromo-N-(6-(4-chlorophenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (15d).
Off white solid; 84% yield; mp 231–233 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.44 (s, 1H), 8.78 (d, J = 7.5 Hz, 2H), 8.44 (dd, J = 8.5, 1.0 Hz, 1H), 7.87–7.81 (m, 2H), 7.60–7.58 (m, 2H), 7.48–7.45 (m, 2H), 4.23 (s, 2H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 169.4, 164.1, 163.9, 149.6, 136.7, 136.2, 136.0, 134.5, 134.0, 131.5, 130.8, 129.7, 129.2, 129.0, 128.2, 121.2, 120.1, 25.1.
4.6.2.5. Spectral data of N-(6-(4-acetylphenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-2-bromoacetamide (15e).
Off white solid; 85% yield; mp 235–237 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.29 (s, 1H), 8.76 (dd, J = 12.0, 4.2 Hz, 2H), 8.32 (dd, J = 8.5, 0.8 Hz, 1H), 8.23 (d, J = 8.3 Hz, 2H), 7.85–7.81 (m, 2H), 7.67 (d, J = 8.3 Hz, 2H), 4.22 (s, 2H), 2.84 (s, 3H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 203.2, 168.2, 163.1, 162.9, 147.9, 144.3, 136.4, 134.7, 133.9, 133.1, 130.5, 130.2, 129.6, 128.8, 128.5, 128.0, 121.4, 120.8, 26.7, 25.4.
4.6.2.6. Spectral data of 2-bromo-N-(1,3-dioxo-6-(thiophen-2-yl)-1H-benzo[de]isoquinolin-2(3H)-yl)acetamide (15f).
Beige solid; 79% yield; mp 225–227 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.17 (s, 1H), 8.75 (dd, J = 8.5, 0.9 Hz, 1H), 8.68 (dd, J = 7.3, 0.9 Hz, 1H), 8.63 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.83–7.79 (m, 1H), 7.61 (dd, J = 5.1, 1.0 Hz, 1H), 7.36 (dd, J = 3.5, 1.0 Hz, 1H), 7.28 (dd, J = 5.1, 3.6 Hz, 1H), 4.21 (s, 2H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 167.6, 162.8, 162.6, 141.8, 139.0, 134.8, 133.5, 132.8, 130.2, 129.7, 129.0, 128.9, 128.6, 128.3, 127.7, 121.3, 120.0, 25.5.
4.7. General procedure for a 6-aryl substituted naphthalimide tethered dihydrofurocoumarin analogue (16a–f)
A reaction mixture containing 15a–f (2.4 mmol), coumarin 5 (389 mg, 2.4 mmol) and benzaldehyde 6a (255 mg, 2.4 mmol) in pyridine (5 mL) was stirred at 80 °C for 7–8 h. After completion of the reaction, cold water was added to the reaction mixture, and the residue obtained was filtered, titrurated well with methanol and vacuum-dried. The crude was purified using column chromatography (CHCl3:EtOAc 90:10) to yield the pure analogues.
4.7.1. Spectral data of N-(1,3-dioxo-6-phenyl-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (16a).
Off white solid; 83% yield; mp 275–277 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.49 (s, 1H), 8.67 (dd, J = 7.3, 2.6 Hz, 1H), 8.64 (dd, J = 7.3, 3.6 Hz, 1H), 8.33 (d, J = 8.5 Hz, 1H), 7.96 (dd, J = 7.8, 1.0 Hz, 1H), 7.92 (td, J = 7.9, 5.0 Hz, 1H), 7.87 (dd, J = 7.5, 3.8 Hz, 1H), 7.80–7.76 (m, 1H), 7.64–7.54 (m, 6H), 7.51 (t, J = 7.6 Hz, 1H), 7.44–7.40 (m, 4H), 7.37–7.34 (m, 1H), 5.72 (d, J = 5.7 Hz, 1H), 4.87 (d, J = 5.7 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.4, 166.1, 161.4, 161.2, 158.3, 154.6, 147.1, 139.6, 137.9, 133.4, 133.2, 131.8, 131.7, 131.4, 131.3, 129.7, 129.5, 128.8, 128.7, 128.6, 128.2, 127.8, 127.7, 127.6, 127.5, 124.4, 123.5, 121.8, 121.7, 120.7, 120.6, 116.7, 111.6, 104.3, 89.0, 50.7; HRMS (ESI): calcd for C36H22N2O6 [M + H]+, 579.1511, found, 579.1556.
4.7.2. Spectral data of N-(6-(4-fluorophenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (16b).
Off white solid; 75% yield; mp 281–283 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.47 (s, 1H), 8.65 (dd, J = 19.0, 7.6 Hz, 2H), 8.31 (d, J = 8.5 Hz, 1H), 7.97–7.91 (m, 2H), 7.88 (dd, J = 7.5, 3.9 Hz, 1H), 7.80–7.76 (m, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 7.3 Hz, 2H), 7.55 (d, J = 8.4 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.43–7.34 (m, 5H), 5.71 (d, J = 5.7 Hz, 1H), 4.85 (d, J = 5.6 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.4, 166.1, 161.4, 161.1, 160.9, 158.3, 154.6, 146.0, 139.6, 134.3, 133.4, 133.1, 132.0, 131.9, 131.8, 131.7, 131.3, 130.4, 129.6, 128.8, 128.4, 127.8, 127.7, 127.6, 127.5, 124.4, 123.5, 121.7, 120.7, 116.6, 115.8, 115.6, 111.6, 104.3, 89.0, 50.7; HRMS (ESI): calcd for C36H21FN2O6 [M + H]+, 597.1417, found, 597.1465.
4.7.3. Spectral data of N-(6-(4-methoxyphenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (16c).
Light yellow solid; 88% yield; mp 285–288 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 11.47 (s, 1H), 8.63 (dt, J = 19.0, 7.4 Hz, 2H), 8.38 (d, J = 8.4 Hz, 1H), 7.97–7.90 (m, 2H), 7.84 (dd, J = 7.6, 3.9 Hz, 1H), 7.78 (t, J = 7.9 Hz, 1H), 7.55 (d, J = 7.9 Hz, 3H), 7.50 (t, J = 7.6 Hz, 1H), 7.43–7.34 (m, 5H), 7.18 (d, J = 8.5 Hz, 2H), 5.68 (d, J = 5.6 Hz, 1H), 4.84 (d, J = 5.7 Hz, 1H), 3.87 (s, 1H); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 167.5, 166.2, 161.4, 161.2, 159.6, 158.3, 154.6, 146.9, 139.7, 133.4, 131.6, 131.4, 131.3, 131.2, 130.1, 129.6, 128.8, 128.1, 127.9, 127.6, 127.5, 125.6, 124.4, 123.5, 121.8, 121.2, 120.2, 116.6, 114.3, 111.7, 104.3, 89.2, 55.3, 50.6; HRMS (ESI): calcd for C37H24N2O7 [M + H]+, 609.1617, found, 609.1657.
4.7.4. Spectral data of N-(6-(4-chlorophenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (16d).
Off white solid; 61% yield; mp 272–274 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.59 (s, 1H), 8.78 (d, J = 4.7 Hz, 2H), 8.46 (dd, J = 8.5, 0.8 Hz, 1H), 7.97 (dd, J = 7.9, 1.3 Hz, 1H), 7.89–7.80 (m, 3H), 7.61–7.58 (m, 2H), 7.54 (dd, J = 16.6, 8.2 Hz, 2H), 7.48–7.46 (m, 2H), 7.40–7.33 (m, 5H), 5.78 (d, J = 6.7 Hz, 1H), 5.18 (d, J = 6.7 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 170.3, 168.7, 161.2, 154.9, 149.4, 137.5, 136.1, 135.7, 134.7, 134.1, 133.6, 130.9, 130.4, 129.4, 129.2, 128.8, 128.7, 128.5, 127.7, 127.3, 125.8, 123.1, 120.6, 119.5, 117.5, 111.4, 104.9, 90.8, 50.7; HRMS (ESI): calcd for C36H21ClN2O6 [M + H]+, 613.1122, found, 613.1166.
4.7.5. Spectral data of N-(6-(4-acetylphenyl)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (16e).
Off white solid; 70% yield; mp 279–281 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.39 (s, 1H), 8.76 (t, J = 7.1 Hz, 2H), 8.33 (dd, J = 8.5, 0.9 Hz, 1H), 8.23 (d, J = 8.4 Hz, 2H), 7.94 (dd, J = 7.9, 1.2 Hz, 1H), 7.86–7.82 (m, 2H), 7.76 (dd, J = 11.6, 4.3 Hz, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.54–7.48 (m, 2H), 7.39–7.32 (m, 5H), 5.72 (d, J = 6.5 Hz, 1H), 5.16 (d, J = 6.5 Hz, 1H), 2.84 (s, 3H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 203.4, 169.8, 168.0, 154.9, 147.9, 144.2, 137.9, 136.2, 134.7, 134.4, 133.9, 133.1, 130.4, 130.1, 129.5, 129.4, 128.7, 128.6, 128.4, 127.9, 127.4, 125.6, 123.1, 121.1, 120.5, 117.5, 111.5, 105.1, 90.7, 50.7, 26.5; HRMS (ESI): calcd for C36H22N2O6 [M + H]+, 621.1617, found, 621.1624.
4.7.6. Spectral data of N-(1,3-dioxo-6-(thiophen-2-yl)-1H-benzo[de]isoquinolin-2(3H)-yl)-4-oxo-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromene-2-carboxamide (16f).
Yellow solid; 73% yield; mp 271–273 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 9.49 (s, 1H), 8.86 (dd, J = 8.5, 0.9 Hz, 1H), 8.73 (dd, J = 26.0, 6.3 Hz, 2H), 7.95 (d, J = 7.8 Hz, 2H), 7.89 (dd, J = 8.5, 7.5 Hz, 1H), 7.79–7.76 (m, 1H), 7.65 (dd, J = 5.1, 1.1 Hz, 1H), 7.52 (dd, J = 10.2, 8.3 Hz, 2H), 7.41 (dd, J = 3.6, 1.1 Hz, 1H), 7.38–7.34 (m, 3H), 7.33–7.29 (m, 3H), 5.74 (d, J = 6.6 Hz, 1H), 5.16 (d, J = 6.7 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 170.0, 168.4, 163.5, 154.9, 142.8, 138.7, 137.7, 135.7, 134.5, 134.1, 133.4, 130.3, 129.9, 129.4, 129.1, 128.9, 128.7, 128.3, 127.8, 127.3, 125.7, 123.1, 120.7, 119.2, 117.5, 111.4, 105.0, 90.8, 50.7; HRMS (ESI): calcd For C34H20N2O6S [M + H]+, 585.1076, found, 585.1079.
4.8. General procedure for the synthesis of intermediates
4.8.1. Synthesis of 3-acetyl-2H-chromen-2-one (19)83.
A mixture of analogue 17 (611 mg, 5 mmol), analogue 18 (651 mg, 5 mmol), and a few drops of piperidine was stirred at room temperature under solvent-free conditions. Precipitation occurred almost immediately, and the reaction progress was tracked using TLC. Upon completion of the reaction, the reaction mixture was poured into crushed ice. The resulting precipitates were collected by filtration, vacuum-dried, and washed with cold MeOH to obtain white solid product 19 in 87% yield; mp 120–122 °C.
4.8.2. Synthesis of 3-(2-bromoacetyl)-2H-chromen-2-one (20)84.
To a stirred solution of 19 (1880 mg, 10 mmol) in a CHCl3–CH3CN mixture (25:5 mL), N-bromosuccinimide (1958 mg, 11 mmol) and p-TSA·H2O (190 mg, 1 mmol) were added, and the reaction was refluxed for 12 h. Once completed, the mixture was cooled, filtered, rinsed with cold MeOH, and dried under vacuum to yield the white solid product 20 in 95% yield; mp 166–168 °C.
4.9. General procedure for synthesis of coumarin tethered dihydrofurocoumarins yielding bis-coumarins (21a–l)
A reaction mixture containing 20 (1 g, 3.76 mmol), coumarin 5 (609 mg, 3.76 mmol), and various aldehydes 6a–l (3.76 mmol) in 5 mL of pyridine was stirred at 80 °C for 6–7 h. Once the reaction was complete, cold water was added, resulting in the precipitation of solid. The obtained residue was filtered, dried well under vacuum and further triturated with methanol to afford the crude product, which was then subjected to column chromatography (CHCl3:EtOAc, 95:5) to yield the pure analogue 21a-l.
4.9.1. Spectral data of 2-(2-oxo-2H-chromene-3-carbonyl)-3-phenyl-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21a).
Off white solid; 85% yield; mp 240–243 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.13 (s, 1H), 7.97 (dd, J = 8.1, 1.4 Hz, 1H), 7.78–7.74 (m, 2H), 7.54 (dd, J = 7.8, 1.2 Hz, 1H), 7.50 (dd, J = 7.7, 6.5 Hz, 2H), 7.44–7.37 (m, 2H), 7.09 (d, J = 10.9 Hz, 1H), 7.04 (t, J = 6.0 Hz, 3H), 7.00 (d, J = 8.1 Hz, 1H), 6.92 (t, J = 7.0 Hz, 1H), 5.45 (d, J = 10.9 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 192.4, 169.3, 163.3, 154.9, 154.8, 150.1, 136.4, 135.1, 134.2, 130.8, 128.8, 128.6, 128.5, 126.1, 125.5, 123.5, 120.8, 117.4, 116.9, 111.7, 104.7, 93.5, 48.5; HRMS (ESI): calcd for C27H16O6 [M + H]+, 437.0980, found, 437.1024.
4.9.2. Spectral data of 2-(2-oxo-2H-chromene-3-carbonyl)-3-(p-tolyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21b).
Cream solid; 88% yield; mp 241–244 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.89 (s, 1H), 7.97 (dd, J = 8.2, 1.3 Hz, 1H), 7.86–7.83 (m, 1H), 7.81 (dd, J = 7.8, 1.3 Hz, 1H), 7.77–7.73 (m, 1H), 7.52–7.48 (m, 4H), 7.15 (s, 4H), 6.67 (d, J = 4.3 Hz, 1H), 4.73 (d, J = 4.3 Hz, 1H), 2.32 (s, 3H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 191.9, 170.1, 164.5, 155.5, 155.3, 153.6, 139.0, 137.3, 136.1, 134.7, 131.5, 130.2, 127.4, 126.9, 126.0, 123.9, 120.5, 118.6, 117.8, 117.5, 112.1, 105.8, 95.3, 48.9, 21.0; HRMS (ESI): calcd for C28H18O6 [M + H]+, 451.1137, found, 451.1180.
4.9.3. Spectral data of 3-(2-chlorophenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21c).
White solid; 79% yield; mp 240–243 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.94 (s, 1H), 7.97 (dd, J = 8.1, 1.5 Hz, 1H), 7.88–7.83 (m, 2H), 7.79–7.76 (m, 1H), 7.54–7.49 (m, 4H), 7.42–7.39 (m, 1H), 7.28–7.26 (m, 2H), 7.21–7.19 (m, 1H), 6.78 (d, J = 5.0 Hz, 1H), 5.36 (d, J = 5.0 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 191.3, 170.3, 163.8, 155.1, 154.9, 153.3, 137.0, 136.1, 134.5, 133.2, 131.1, 130.1, 129.7, 128.3, 128.0, 126.5, 125.7, 123.5, 120.2, 118.2, 117.4, 117.2, 111.6, 93.9, 45.3; HRMS (ESI): calcd for C27H15ClO6 [M + H]+, 471.0557, found, 471.0632.
4.9.4. Spectral data of 3-(4-chlorophenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21d).
White solid; 57% yield; mp 247–249 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm); 8.87 (s, 1H), 7.95 (dd, J = 8.1, 1.6 Hz, 1H), 7.86–7.80 (m, 2H), 7.77–7.73 (m, 1H), 7.52–7.47 (m, 4H), 7.33–7.31 (m, 2H), 7.24–7.21 (m, 2H), 6.63 (d, J = 4.1 Hz, 1H), 4.75 (d, J = 4.1 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 190.7, 169.5, 163.6, 155.2, 154.9, 153.0, 137.4, 136.8, 134.4, 131.1, 129.3, 128.6, 126.4, 125.6, 123.6, 120.2, 118.2, 117.4, 117.2, 111.7, 105.1, 94.5, 48.0; HRMS (ESI): calcd for C27H15ClO6 [M + H]+, 471.0557, found, 471.0634.
4.9.5. Spectral data of 3-(3-nitrophenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21e).
Off white solid; 83% yield; mp 253–255 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.89 (s, 1H), 8.20–8.18 (m, 2H), 7.97 (dd, J = 7.9, 1.3 Hz, 1H), 7.85–7.80 (m, 2H), 7.77–7.74 (m, 1H), 7.72–7.69 (m, 1H), 7.56 (dd, J = 8.8, 7.7 Hz, 1H), 7.51–7.47 (m, 4H), 6.66 (d, J = 4.1 Hz, 1H), 4.88 (d, J = 4.1 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 189.7, 169.7, 162.9, 155.3, 155.1, 152.9, 148.5, 141.4, 136.8, 134.5, 134.0, 131.1, 130.1, 126.4, 125.6, 123.7, 123.3, 122.3, 120.1, 118.2, 117.5, 117.2, 111.6, 104.6, 94.0, 48.2; HRMS (ESI): calcd for C27H15NO8 [M + H]+, 482.0831, found, 482.0874.
4.9.6. Spectral data of 3-(4-nitrophenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21f).
Off white solid; 41% yield; mp 254–256 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.97 (s, 1H), 8.27–8.24 (m, 2H), 8.00 (dd, J = 6.5, 1.4 Hz, 1H), 7.90–7.85 (m, 2H), 7.82–7.78 (m, 1H), 7.57–7.51 (m, 6H), 6.70 (d, J = 4.1 Hz, 1H), 4.92 (d, J = 4.1 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 190.1, 169.9, 163.4, 155.2, 155.0, 153.6, 147.4, 146.7, 137.1, 134.7, 131.2, 128.4, 126.7, 126.6, 125.8, 124.4, 124.0, 123.6, 119.8, 118.2, 117.5, 117.2, 111.5, 104.5, 94.0, 48.2; HRMS (ESI): calcd for C27H15NO8 [M + H]+, 482.0831, found, 482.0872.
4.9.7. Spectral data of 3-(2-methoxyphenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21g).
Light yellow solid; 93% yield; mp 256–258 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.82 (s, 1H), 7.95 (dd, J = 7.9, 1.4 Hz, 1H), 7.85–7.81 (m, 2H), 7.77–7.73 (m, 1H), 7.52–7.48 (m, 4H), 7.31–7.26 (m, 1H), 7.05 (dd, J = 7.6, 1.6 Hz, 1H), 6.94–6.88 (m, 2H), 6.72 (d, J = 5.1 Hz, 1H), 5.14 (d, J = 5.0 Hz, 1H), 3.61 (s, 3H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 192.3, 170.3, 164.0, 156.4, 155.0, 154.9, 152.1, 136.5, 134.2, 131.0, 129.7, 127.8, 126.5, 126.4, 125.6, 123.5, 121.4, 121.0, 118.2, 117.4, 117.0, 111.8, 111.2, 103.7, 93.8, 55.3, 42.9; HRMS (ESI): calcd for C28H18O7 [M + H]+, 467.1086, found, 467.1128.
4.9.8. Spectral data of 3-(4-methoxyphenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21h).
Off white solid; 95% yield; mp 265–267 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.88 (s, 1H), 7.96 (d, J = 7.6 Hz, 1H), 7.83 (dd, J = 18.7, 7.9 Hz, 2H), 7.75 (t, J = 7.7 Hz, 1H), 7.50 (dd, J = 11.2, 8.0 Hz, 4H), 7.22 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 6.64 (d, J = 4.0 Hz, 1H), 4.73 (d, J = 3.9 Hz, 1H), 3.86 (s, 3H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 191.1, 169.5, 163.9, 158.5, 155.1, 154.9, 153.0, 136.8, 134.3, 131.9, 131.1, 128.5, 126.4, 125.6, 123.5, 120.2, 118.2, 117.4, 117.1, 114.8, 111.8, 105.4, 94.8, 55.6, 48.1; HRMS (ESI): calcd for C28H18O7 [M + H]+, 467.1086, found, 467.1130.
4.9.9. Spectral data of 3-(4-oxo-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-3-yl)benzonitrile (21i).
White solid; 76% yield; mp 271–273 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.94 (s, 1H), 7.98 (dd, J = 7.8, 1.2 Hz, 1H), 7.88–7.83 (m, 2H), 7.77 (dd, J = 11.6, 4.4 Hz, 1H), 7.69–7.65 (m, 3H), 7.54–7.49 (m, 5H), 6.64 (d, J = 4.0 Hz, 1H), 4.82 (d, J = 3.9 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 189.9, 169.9, 163.3, 155.2, 155.0, 153.4, 141.0, 137.0, 134.6, 133.0, 132.2, 131.2, 131.1, 130.1, 126.6, 125.7, 123.7, 119.8, 118.2, 117.5, 117.2, 111.6, 111.5, 104.5, 94.1, 48.1; HRMS (ESI): calcd for C28H15NO6 [M + H]+, 462.0933, found, 462.0976.
4.10.10. Spectral data of 3-(2,5-dimethoxyphenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21j).
Light yellow solid; 85% yield; mp 258–260 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.85 (s, 1H), 7.95 (dd, J = 7.8, 1.4 Hz, 1H), 7.84 (t, J = 7.9 Hz, 2H), 7.78–7.74 (m, 1H), 7.52 (dd, J = 7.5, 1.2 Hz, 2H), 7.50–7.48 (m, 2H), 6.89 (dd, J = 9.0, 2.8 Hz, 1H), 6.86 (d, J = 9.0 Hz, 1H), 6.75 (d, J = 2.8 Hz, 1H), 6.73 (d, J = 5.1 Hz, 1H), 5.11 (d, J = 5.0 Hz, 1H), 3.79 (s, 3H), 3.60 (s, 3H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 192.4, 170.8, 164.4, 155.3, 155.2, 153.1, 152.6, 151.8, 136.9, 134.6, 131.3, 128.4, 126.7, 125.9, 123.8, 121.1, 118.5, 117.7, 117.3, 114.4, 112.7, 112.0, 103.7, 94.0, 56.7, 56.2, 43.2; HRMS (ESI): calcd for C29H20O8 [M + H]+, 497.1192, found, 497.1234.
4.10.11. Spectral data of 3-(2,3-dichlorophenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21k).
White solid; 71% yield; mp 271–273 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.98 (s, 1H), 7.96 (dd, J = 8.1, 1.6 Hz, 1H), 7.89–7.85 (m, 2H), 7.81–7.77 (m, 1H), 7.56–7.51 (m, 5H), 7.47 (dd, J = 8.0, 1.5 Hz, 1H), 7.22 (t, J = 7.9 Hz, 1H), 7.14 (dd, J = 7.8, 1.5 Hz, 1H), 6.76 (d, J = 4.9 Hz, 1H), 5.43 (d, J = 4.4 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 191.0, 170.3, 155.1, 155.0, 153.7, 137.1, 134.6, 131.7, 131.2, 130.5, 128.1, 126.6, 125.8, 123.5, 119.9, 118.2, 117.4, 117.2, 111.5, 91.4, 45.3; HRMS (ESI): calcd for C27H14Cl2O6 [M + H]+, 505.0167, found, 505.0247.
4.10.12. Spectral data of 3-(4-bromophenyl)-2-(2-oxo-2H-chromene-3-carbonyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (21l).
Off white solid; 83% yield; mp 281–283 °C; 1H NMR (CDCl3 + TFA, 500 MHz): δ (ppm) 8.80 (s, 1H), 7.93 (dd, J = 8.2, 1.5 Hz, 1H), 7.82–7.76 (m, 2H), 7.74–7.70 (m, 1H), 7.49–7.44 (m, 6H), 7.19–7.16 (m, 2H), 6.59 (d, J = 4.0 Hz, 1H), 4.72 (d, J = 4.0 Hz, 1H); 13C NMR (CDCl3 + TFA, 125 MHz): δ (ppm) 190.2, 169.2, 163.0, 155.3, 155.0, 152.3, 138.1, 136.5, 134.1, 132.2, 131.0, 129.0, 126.2, 125.4, 123.6, 122.3, 120.5, 118.2, 117.4, 117.2, 111.8, 105.1, 94.4, 48.0; HRMS (ESI): calcd for C27H15BrO6 [M + H]+, 515.0052, found, 515.0126.
Author contributions
A. J.: conceptualization, methodology, and drafting; K. P.: visualization, investigation, supervision and editing.
Conflicts of interest
The authors declare no competing financial interest.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: materials and methods; additional tables and figures of related biological studies; and spectral and other characterization data for key target analogues. See DOI: https://doi.org/10.1039/d5tb01511a.
Acknowledgements
KP thanks CEEMS (Project No: TIET/CEEMS/Regular/2021/018/8037), ANRF (SERB) (CRG/2023/004080) for financial support, SAIF-PU for NMR analysis and DST-FIST (SR/FST/CS-II/2018/69) for HRMS analysis.
References
- C. S. Ho, C. T. H. Wong, T. T. Aung, R. Lakshminarayanan, J. S. Mehta, S. Rauz, A. McNally, B. Kintses, S. J. Peacock, C. de la Fuente-Nunez, R. E. W. Hancock and D. S. J. Ting, Antimicrobial resistance: a concise update, Lancet Microbe, 2025, 6, 100947 CrossRef.
- C. Wang, X. Wei, L. Zhong, C. L. Chan, H. Li and H. Sun, Metal-Based Approaches for the Fight against Antimicrobial Resistance: Mechanisms, Opportunities, and Challenges, J. Am. Chem. Soc., 2025, 147, 12361–12380 CrossRef.
- J. Gao, H. Hou and F. Gao, Current scenario of quinolone hybrids with potential antibacterial activity against ESKAPE pathogens, Eur. J. Med. Chem., 2023, 247, 115026 CrossRef PubMed.
- P. Barathe, K. Kaur, S. Reddy, V. Shriram and V. Kumar, Antibiotic pollution and associated antimicrobial resistance in the environment, J. Hazard. Mater. Lett., 2024, 5, 100105 CrossRef.
- C. R. MacNair, S. T. Rutherford and M. W. Tan, Alternative therapeutic strategies to treat antibiotic-resistant pathogens, Nat. Rev. Microbiol., 2024, 22, 262–275 CrossRef.
- N. Parvin, S. W. Joo and T. K. Mandal, Nanomaterial-Based Strategies to Combat Antibiotic Resistance: Mechanisms and Applications, Antibiotics, 2025, 14, 207 CrossRef.
- S. K. Verma, S. Rangappa, R. Verma, F. Xue, S. Verma, K. S. S. Kumar and K. S. Rangappa, Sulfur (SVI)-containing heterocyclic hybrids as antibacterial agents against methicillin-resistant Staphylococcus Aureus (MRSA) and its SAR, Bioorg. Chem., 2024, 145, 107241 CrossRef PubMed.
- K. B. Gangurde, R. A. More, V. A. Adole and D. S. Ghotekar, Design, synthesis and biological evaluation of new series of benzotriazole-pyrazole clubbed thiazole hybrids as bioactive heterocycles: Antibacterial, antifungal, antioxidant, cytotoxicity Study, J. Mol. Struct., 2024, 1299, 136760 CrossRef.
- F. Annunziata, C. Pinna, S. Dallavalle, L. Tamborini and A. Pinto, An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities, Int. J. Mol. Sci., 2020, 21, 4618 CrossRef PubMed.
- X. C. Yang, C. M. Zeng, S. R. Avula, X. M. Peng, R. X. Geng and C. H. Zhou, Novel coumarin aminophosphonates as potential multitargeting antibacterial agents against Staphylococcus aureus, Eur. J. Med. Chem., 2023, 245, 114891 CrossRef PubMed.
- X. C. Yang, P. L. Zhang, K. V. Kumar, S. Li, R. X. Geng and C. H. Zhou, Discovery of unique thiazolidinone-conjugated coumarins as novel broad spectrum antibacterial agents, Eur. J. Med. Chem., 2022, 232, 114192 CrossRef PubMed.
- F. Salehian, H. Nadri, L. Jalili-Baleh, L. Youseftabar-Miri, S. N. A. Bukhari, A. Foroumadi, T. T. Küçükkilinç, M. Sharifzadeh and M. Khoobi, A review: Biologically active 3,4-heterocycle-fused coumarins, Eur. J. Med. Chem., 2021, 212, 113034 CrossRef CAS PubMed.
- Y. Hu, Y. Shen, X. Wu, X. Tu and G. X. Wang, Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents, Eur. J. Med. Chem., 2018, 143, 958–969 CrossRef CAS.
- X. C. Yang, C. F. Hu, P. L. Zhang, S. Li, C. S. Hu, R. X. Geng and C. H. Zhou, Coumarin thiazoles as unique structural skeleton of potential antimicrobial agents, Bioorg. Chem., 2022, 124, 105855 CrossRef CAS.
- C. G. Arya, R. Kishore, P. Gupta, R. Gondru, J. Arockiaraj, M. Pasupuleti, M. Chandrakanth, V. P. Punya and J. Banothu, Identification of coumarin – benzimidazole hybrids as potential antibacterial agents: Synthesis, in vitro and in vivo biological assessment, and ADMET prediction, Bioorg. Med. Chem.
Lett., 2024, 110, 129881 CrossRef CAS PubMed.
- S. Rohilla, S. Shah and V. K. Singh, Copper-Catalyzed Asymmetric Propargylic [3 + 2] Cycloaddition: Synthesis of Enantioenriched Dihydrofuro[3,2-c]coumarins and its Quinolinone and Thiocoumarin Analogues, Org. Lett., 2023, 25, 3733–3738 CrossRef CAS PubMed.
- S. Dey, A. Das, R. N. Yadav, P. J. Boruah, K. Sarkar, A. K. Paul and M. F. Hossain, Electron donor-acceptor complex enabled photocascade strategy for the synthesis of trans-dihydrofuro[3,2-c]chromen-4-one scaffolds via radical conjugate addition of pyridinium ylide, Chem. Commun., 2024, 60, 14384–14387 RSC.
- G. Mali, S. Maji, K. A. Chavan, M. Shukla, M. Kumar, S. Bhattacharyya and R. D. Erande, Effective Synthesis and Biological Evaluation of Functionalized 2,3-Dihydrofuro[3,2-c]coumarins via an Imidazole-Catalyzed Green Multicomponent Approach, ACS Omega, 2022, 7, 36028–36036 CrossRef CAS PubMed.
- Z. Liu, Q. Liu, L. Chen, Y. Liu, T. Zhao, L. Yang, M. Zhang and C. Yao, Novel indolespiro bicoumarins: synthesis and their antifungal activity, Org. Biomol. Chem., 2025, 23, 4960–4965 RSC.
- U. Salar, A. Nizamani, F. Arshad, K. M. Khan, M. I. Fakhri, S. Perveen, N. Ahmed and M. I. Choudhary, Bis-coumarins; non-cytotoxic selective urease inhibitors and antiglycation agents, Bioorg. Chem., 2019, 91, 103170 CrossRef PubMed.
- S. Walki, G. H. Malimath, K. M. Mahadevan, S. Naik, S. M. Sutar, H. Savanur and L. Naik, Synthesis, spectroscopic properties, and DFT correlative studies of 3,3′- carbonyl biscoumarin derivatives, J. Mol. Struct., 2021, 1243, 130781 CrossRef CAS.
- L. L. Mou, X. M. Wu, A. Bibi, J. X. Wang and C. H. Zhou, A comprehensive insight into naphthalimides as novel structural skeleton of multitargeting promising antibiotics, Future Med. Chem., 2025, 17, 575–590 CrossRef CAS.
- P. L. Zhang, M. H. Laiche, Y. L. Li, W. W. Gao, J. M. Lin and C. H. Zhou, An unanticipated discovery of novel naphthalimidopropanediols as potential broad-spectrum antibacterial members, Eur. J. Med. Chem., 2022, 241, 114657 CrossRef CAS.
- T. Wang, Y. Gao, F. Wu, L. Luo, J. Ma and Y. Hu, Coumarin-furo[2,3-d]pyrimidone hybrid molecules targeting human liver cancer cells: synthesis, anticancer effect, EGFR inhibition and molecular docking studies, RSC Med. Chem., 2024, 15, 1565–1577 RSC.
- P. P. Navarro, A. Vettiger, V. Y. Ananda, P. M. Llopis, C. Allolio, T. G. Bernhardt and L. H. Chao, Cell wall synthesis and remodelling dynamics determine division site architecture and cell shape in Escherichia coli, Nat. Microbiol., 2022, 7, 1621–1634 CrossRef CAS PubMed.
- A. Naskar, J. Shin and K. S. Kim, A MoS2 based silver-doped ZnO nanocomposite and its antibacterial activity against β-lactamase expressing Escherichia coli, RSC Adv., 2022, 12, 7268–7275 RSC.
- D. Koszelewski, P. Kowalczyk, A. Brodzka, A. Hrunyk, K. Kramkowski and R. Ostaszewski, Enzymatic Synthesis of a Novel Coumarin Aminophosphonates: Antibacterial Effects and Oxidative Stress Modulation on Selected E. coli Strains, Int. J. Mol. Sci., 2023, 24, 7609 CrossRef CAS PubMed.
- A. Fenollar-Penadés, P. Catalá-Gregori, V. Tallá-Ferrer, M. Á. Castillo, M. García-Ferrús and A. Jiménez-Belenguer, Evolution of the Antibiotic Resistance Levels, Multi-Resistance Patterns, and Presence of Antibiotic Resistance Genes in E. coli Isolates from the Feces of Breeding Hens during the Rearing Period, Antibiotics, 2024, 13, 753 CrossRef.
- F. Gao, P. Wang, H. Yang, Q. Miao, L. Ma and G. Lu, Recent developments of quinolone-based derivatives and their activities against Escherichia coli, Eur. J. Med. Chem., 2018, 157, 1223–1248 CrossRef CAS PubMed.
- S. Fang, W. T. Kang, H. Li, Q. Cai, W. Liang, M. Zeng, Q. Yu, R. Zhong, Y. Tao, S. Liu and S. Lin, Development of cannabidiol derivatives as potent broad-spectrum antibacterial agents with membrane-disruptive mechanism, Eur. J. Med. Chem., 2024, 266, 116149 CrossRef CAS PubMed.
- S. Gupta and K. Paul, Membrane-active substituted triazines as antibacterial agents against Staphylococcus aureus with potential for low drug resistance and broad activity, Eur. J. Med. Chem., 2023, 258, 115551 CrossRef CAS PubMed.
- A. Kumari and R. K. Singh, Morpholine as ubiquitous pharmacophore in medicinal chemistry: Deep insight into the structure-activity relationship (SAR), Bioorg. Chem., 2020, 96, 103578 CrossRef CAS.
- Y. M. Tan, D. Li, F. F. Li, M. Fawad Ansari, B. Fang and C. H. Zhou, Pyrimidine-conjugated fluoroquinolones as new potential broad-spectrum antibacterial agents, Bioorg. Med. Chem. Lett., 2022, 73, 128885 CrossRef CAS PubMed.
- N. Sheikhi, M. Bahraminejad, M. Saeedi and S. S. Mirfazli, A review: FDA-approved fluorine-containing small molecules from 2015 to 2022, Eur. J. Med. Chem., 2023, 260, 115758 CrossRef CAS PubMed.
- A. Khanna, N. Kumar, R. Rana, Jyoti, A. Sharma, Muskan, H. Kaur and P. M. S. Bedi, Fluoroquinolones tackling antimicrobial resistance: Rational design, mechanistic insights and comparative analysis of norfloxacin vs ciprofloxacin derivatives, Bioorg. Chem., 2024, 153, 107773 CrossRef CAS PubMed.
- J. X. Wang, P. L. Zhang, L. Gopala, J. S. Lv, J. M. Lin and C. H. Zhou, A Unique Hybridization Route to Access Hydrazylnaphthalimidols as Novel Structural Scaffolds of Multitargeting Broad-Spectrum Antifungal Candidates, J. Med. Chem., 2024, 67, 8932–8961 CrossRef CAS.
- Y. M. Tan, J. Zhang, Y. J. Wei, Y. G. Hu, S. R. Li, S. L. Zhang and C. H. Zhou, Cyanomethylquinolones as a New Class of Potential Multitargeting Broad-Spectrum Antibacterial Agents, J. Med. Chem., 2024, 67, 9028–9053 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef CAS PubMed.
- M. S. Raghu, C. B. Pradeep Kumar, K. Yogesh Kumar, M. K. Prashanth, F. Alharethy and B. H. Jeon, Synthesis, biological evaluation and molecular docking study of pyrimidine linked thiazolidinedione derivatives as potential antimicrobial and antitubercular agents, Bioorg. Med. Chem. Lett., 2024, 103, 129707 CrossRef CAS PubMed.
- W. Wang, R. Wang, L. An, L. Li, H. Xiong, D. Li, F. Dong, J. Lei, M. Wang, Z. Yang, H. Wang, X. Ling, C. Fountzilas, F. Li and Q. Li, Design, synthesis and investigation of biological activity and mechanism of fluoroaryl-substituted derivatives at the FL118 position 7, Eur. J. Med. Chem., 2025, 283, 117143 CrossRef CAS PubMed.
- K. Ciesielska, D. Wawrzyniak, G. Dutkiewicz, M. Kubicki, W. Jankowski, M. Hoffmann, K. Kamel, K. Rolle and D. Pluskota-Karwatka, Diastereoselective synthesis and biological evaluation of new fluorine-containing α-aminophosphonates as anticancer agents and scaffold to human urokinase plasminogen activator inhibitors, Eur. J. Med. Chem., 2025, 283, 117116 CrossRef CAS PubMed.
- N. Manju, A. K. Raigar, K. Saini, N. Jyoti, R. K. Kapavarapu and A. Guleria, Spiro thiochromene-oxindoles as novel anti-inflammatory agents: design, sustainable synthesis, in vitro and in silico evaluations, RSC Adv., 2025, 15, 261–275 RSC.
- E. A. Basiony, A. A. Hassan, M. Elsawalhy, A. A. H. Abdel-Rahman, H. Mansour, R. K. Arafa and N. A. Hassan, Rational design, synthesis, biological evaluation, molecular docking, and molecular dynamics of substituted uracil
derivatives as potent anti-cancer agents, Bioorg. Chem., 2025, 154, 108066 CrossRef CAS.
- S. D. Kumar, J. K. Lee, N. K. Radhakrishnan, J. K. Bang, B. Kim, S. C. Chaudhary, A. Chelladurai, B. Ganbaatar, E. Y. Kim, C. W. Lee, S. Yang, Y. Kim and S. Y. Shin, Antibacterial, Antibiofilm, and Anti-Inflammatory Effects of a Novel Thrombin-Derived Peptide in Sepsis Models: Insights into Underlying Mechanisms, J. Med. Chem., 2024, 67, 19791–19812 CrossRef CAS.
- Q. Tang, H. Zhang, K. Chandarajoti, Z. Jiao, L. Nie, S. Lv, J. Zuo, W. Zhou and X. Han, Design and synthesis of coumarin-based amphoteric antimicrobials with biofilm interference and immunoregulation effects, RSC Med. Chem., 2025, 16, 1223–1234 RSC.
- G. E. Ahmed, Z. A. Elshahid, E. R. El-Sawy, M. S. Abdel-Aziz and A. Abdel-Aziem, Synthesis, biofilm formation inhibitory, and inflammation inhibitory activities of new coumarin derivatives, Sci. Rep., 2024, 14, 9106 CrossRef CAS PubMed.
- X. M. Zhou, Y. Y. Hu, B. Fang and C. H. Zhou, Benzenesulfonyl thiazoloimines as unique multitargeting antibacterial agents towards Enterococcus faecalis, Eur. J. Med. Chem., 2023, 248, 115088 CrossRef CAS.
- J. Dai, Q. Li, Z. Li, Z. Zang, Y. Luo and C. Zhou, Discovery of Quinazolone Pyridiniums as Potential Broad-Spectrum Antibacterial Agents, Molecules, 2025, 30, 243 CrossRef CAS.
- J. Zhang, Y. M. Tan, S. R. Li, N. Battini, S. L. Zhang, J. M. Lin and C. H. Zhou, Discovery of benzopyridone cyanoacetates as new type of potential broad-spectrum antibacterial candidates, Eur. J. Med. Chem., 2024, 265, 116107 CrossRef CAS.
- Y. X. Wang, H. R. Wang, J. S. Zhao, X. C. Yang, B. Fang, Z. L. Zang, R. X. Geng and C. H. Zhou, Benzo-α-pyrone-derived multitargeting actions to enhance the antibacterial performance of sulfanilamides against Escherichia coli, Bioorg. Chem., 2025, 158, 108339 CrossRef CAS PubMed.
- M. Niu, X. Gu, J. Yang, H. Cui, X. Hou, Y. Ma, C. Wang and G. Wei, Dual-Mechanism Glycolipidpeptide with High Antimicrobial Activity, Immunomodulatory Activity, and Potential Application for Combined Antibacterial Therapy, ACS Nano, 2023, 17, 6292–6316 CrossRef CAS.
- Y. G. Hu, N. Battini, B. Fang and C. H. Zhou, Discovery of indolylacryloyl-derived oxacins as novel potential broad-spectrum antibacterial candidates, Eur. J. Med. Chem., 2024, 270, 116392 CrossRef CAS.
- W. Zhou, Q. Qiao, Y. Tao, C. Duan, J. Li, X. Fang, N. Xu, J. Chen, W. Liu, L. Miao and Z. Xu, Bacterial membrane-induced disassembly of fluorescein aggregates enables selective imaging and killing of Gram-positive bacteria, Sens. Actuators, B, 2024, 410, 135691 CrossRef CAS.
- Y. P. Xie, N. Sangaraiah, J. P. Meng and C. H. Zhou, Unique Carbazole-Oxadiazole Derivatives as New Potential Antibiotics for Combating Gram-positive and -Negative Bacteria, J. Med. Chem., 2022, 65, 6171–6190 CrossRef CAS.
- W. Dan, Y. Xiong, R. Li, H. Gao, P. Liu, M. Sui, C. Xu and J. Dai, Synthesis and anti-MRSA activity of quaternized small molecule antimicrobial peptide mimics based on norharmane, Eur. J. Med. Chem., 2025, 290, 117518 CrossRef CAS.
- M. Sui, J. Zhang, J. Li, L. Wang, Z. Gao, W. Dan and J. Dai, Antibacterial activity and multi-target mechanism of harmane against Escherichia coli O157:H7 and its application on ready-to-eat leafy greens, Int. J. Food Microbiol., 2025, 431, 111084 CrossRef CAS.
- F. F. Li, W. H. Zhao, V. K. R. Tangadanchu, J. P. Meng and C. H. Zhou, Discovery of novel phenylhydrazone-based oxindole-thiolazoles as potent antibacterial agents toward Pseudomonas aeruginosa, Eur. J. Med. Chem., 2022, 239, 114521 CrossRef CAS.
- H. Sun, S. Y. Huang, P. Jeyakkumar, G. X. Cai, B. Fang and C. H. Zhou, Natural Berberine-Derived Azolyl Ethanols as New Structural Antibacterial Agents against Drug-Resistant Escherichia coli, J. Med. Chem., 2022, 65, 436–459 CrossRef CAS.
- W. Zhang, Y. Ran, M. Yang, Y. Hu, Z. Wang, Y. Cao and H. Ran, An Oxidative Stress Nano-Amplifier for Improved Tumor Elimination and Combined Immunotherapy, Adv. Healthcare Mater., 2024, 13, 2402349 CrossRef CAS.
- H. Niu, S. Wang, Y. Liu, N. Ma, S. Cheng, B. Feng, H. Jeong, Y. Yang, G. Wang, T. D. James, J. Yoon, J. L. Sessler and H. Zhang, Naphthalimide-Based Type-I Nano-Photosensitizers for Enhanced Antitumor Photodynamic Therapy: H2S Synergistically Regulates PeT and Self-Assembly, Angew. Chem., Int. Ed., 2025, e202512150 CAS.
- S. R. Li, C. M. Zeng, X. M. Peng, J. P. Chen, S. Li and C. H. Zhou, Benzopyrone-mediated quinolones as potential multitargeting antibacterial agents, Eur. J. Med. Chem., 2023, 262, 115878 CrossRef PubMed.
- X. Y. Yuan, H. Liu and B. Sun, Construction of Guanidinium-Functionalized Covalent Organic Frameworks via Phototriggered Click Reaction as a Dual-Mode Accurate Sensor for Malondialdehyde, ACS Sens., 2025, 10, 1135–1146 CrossRef CAS PubMed.
- S. Paul, S. Biswas, T. Choudhuri, S. Bandyopadhyay, S. Mandal and A. K. Bagdi, I2-Catalyzed Cascade Annulation/Cross-Dehydrogenative Coupling: Excellent Platform to Access 3-Sulfenyl Pyrazolo[1,5-a]pyrimidines with Potent Antibacterial Activity against Pseudomonas aeruginosa and Staphylococcus aureus, ACS Appl. Bio Mater., 2025, 8, 3254–3269 CrossRef CAS PubMed.
- Y. Wang, F. Huo and C. Yin, Development of Human Serum Albumin Fluorescent Probes in Detection, Imaging, and Disease Therapy, J. Phys. Chem. B, 2024, 128, 1121–1138 CrossRef CAS PubMed.
- S. Gupta, V. Luxami and K. Paul, Bacterial Cell Death to Overcome Drug Resistance with Multitargeting Bis-Naphthalimides as Potent Antibacterial Agents against Enterococcus faecalis, J. Mater. Chem. B, 2024, 12, 5645–5660 RSC.
- P. Vidhyapriya, D. Divya, B. Manimaran and N. Sakthivel, Molecular interaction of manganese based carbon monoxide releasing molecule (MnCORM) with human serum albumin (HSA), Bioorg. Chem., 2019, 92, 103078 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, Spectrophotometry of Iodine with Aromatic Hydrocarbons, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- X. Meng, Z. Xia, J. Cheng, Y. Wang, X. Ren, L. He and D. Liu, Assessment of the binding mechanism of ergothioneine to human serum albumin: Multi-spectroscopy, molecular docking and molecular dynamic simulation, Spectrochim. Acta - Part A Mol. Biomol. Spectrosc., 2025, 327, 125368 CrossRef CAS PubMed.
- M. Zahirul Kabir, H. Tayyab, C. Erkmen, S. B. Mohamad and B. Uslu, Spectroscopic, voltammetric and computational approaches shed light on the combination characteristics of an anticancer agent, bexarotene with human serum albumin, J. Photochem. Photobiol. A Chem., 2023, 443, 114881 CrossRef CAS.
- N. Farajzadeh-Dehkordi, S. Farhadian, Z. Zahraei, S. Asgharzadeh, B. Shareghi and B. Shakerian, Insights into the binding interaction of Reactive Yellow 145 with human serum albumin from a biophysics point of view, J. Mol. Liq., 2023, 369, 120800 CrossRef CAS.
- M. L. da Silva Moreira, O. A. Chaves, N. C. de Lucas, J. da Silva Goulart, S. J. Garden, C. Serpa and J. C. Netto-Ferreira, Spectroscopic and in silico characterization of the interaction between synthetic 2-substituted-naphtho-1,4-quinones and human serum albumin, J. Mol. Liq., 2024, 403, 124829 CrossRef CAS.
- D. Roccatano, Binding dynamics of linear alkyl-sulfates of different chain lengths on a protein surface, J. Mol. Liq., 2024, 414, 126168 CrossRef CAS.
- V. Rupreo, R. Tissopi, K. Baruah, A. S. Roy and J. Bhattacharyya, Multispectroscopic and Theoretical Investigation on the Binding Interaction of a Neurodegenerative Drug, Lobeline with Human Serum Albumin: Perturbation in Protein Conformation and Hydrophobic-Hydrophilic Surface, Mol. Pharm., 2024, 21, 4169–4182 CrossRef CAS.
- W. Wang, Q. Sun, N. Gan, Y. Zhai, H. Xiang and H. Li, Characterizing the interaction between methyl ferulate and human serum albumin by saturation transfer difference NMR, RSC Adv., 2020, 10, 32999–33009 RSC.
- P. Yadav, J. K. Yadav, A. Agarwal and S. K. Awasthi, Insights into the interaction of potent antimicrobial chalcone triazole analogs with human serum albumin: spectroscopy and molecular docking approaches, RSC Adv., 2019, 9, 31969–31978 RSC.
- S. Dhiman, R. Kour, G. Kumar, S. Kaur, V. Luxami, P. Singh and S. Kumar, “Turn-On” monopodal and dipodal nanoprobes for serum albumins - a case of shift in selectivity towards BSA and a Z- to U-like aonformational change, Mater. Chem. Front., 2022, 6, 2651–2660 RSC.
- P. C. Agu, C. A. Afiukwa, O. U. Orji, E. M. Ezeh, I. H. Ofoke, C. O. Ogbu, E. I. Ugwuja and P. M. Aja, Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management, Sci. Rep., 2023, 13, 13398 CrossRef CAS.
- S. R. Li, J. C. Yu, Y. Cheng and C. H. Zhou, Discovery of benzopyronyl imidazolidinediones as new structural skeleton of potential multitargeting antibacterial agents, Bioorg. Chem., 2025, 164, 108923 CrossRef CAS PubMed.
- W. H. Zhao, J. H. Xu, V. K. R. Tangadanchu and C. H. Zhou, Thiazolyl hydrazineylidenyl indolones as unique potential multitargeting broad-spectrum antimicrobial agents, Eur. J. Med. Chem., 2023, 256, 115452 CrossRef CAS.
- H. S. El-Hema, S. M. Soliman, W. El-Dougdoug, M. H. M. Ahmed, A. Abdelmajeid, E. S. Nossier, M. F. Hussein, A. A. Alrayes, M. Hassan, N. A. Ahmed, A. Sabry and A. A. H. Abdel-Rahman, Design, Characterization, Antimicrobial Activity, and In Silico Studies of Theinothiazoloquinazoline Derivatives Bearing Thiazinone, Tetrazole, and Triazole Moieties, ACS Omega, 2025, 10, 9703–9717 CrossRef CAS PubMed.
- S. Gul, A. Alam, Zainab, M. Assad, A. A. Elhenawy, M. S. Islam, S. A. A. Shah, Z. Parveen, T. A. Shah and M. Ahmad, Exploring the synthesis, molecular structure and biological activities of novel Bis-Schiff base derivatives: A combined theoretical and experimental approach, J. Mol. Struct., 2024, 1306, 137828 CrossRef CAS.
- A. Jain and K. Paul, Column Chromatography-Free Synthesis of Spirooxindole and Spiroindanone-Based Naphthalimides as Potent c-MYC G4 Stabilizers and HSA Binders for Elevating Anticancer Potential, ACS Appl. Bio Mater., 2025, 8, 3728–3747 CrossRef CAS.
- A. Sethi, K. Sasikala, P. Jakkula, D. Gadde, S. Sanam, I. A. Qureshi, V. Talla and M. Alvala, Design, synthesis and computational studies involving indole-coumarin hybrids as galectin-1 inhibitors, Chem. Pap., 2021, 75, 2791–2805 CrossRef CAS.
- B. H. Naguib, H. A. Elsebaie, M. S. Nafie, S. Mohamady, N. R. Albujuq, A. Samir Ayed, D. Nada, A. F. Khalil, S. M. Hefny, H. O. Tawfik and M. A. Shaldam, Fragment-based design and synthesis of coumarin-based thiazoles as dual c-MET/STAT-3 inhibitors for potential antitumor agents, Bioorg. Chem., 2024, 151, 107682 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab