
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT investigation of active sites in metal-doped ferrierite zeolites for selective methane oxidation to methanol
Zirong Guan†
 a,
Jiaming Miao†a,
Wenfu Yana,
Zexing Qu*b and
Yi Li*ac
a,
Jiaming Miao†a,
Wenfu Yana,
Zexing Qu*b and
Yi Li*ac
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China. E-mail: yili@jlu.edu.cn
bInstitute of Theoretical Chemistry, College of Chemistry, Jilin University, Changchun, 130023, P. R. China. E-mail: zxqu@jlu.edu.cn
cInternational Center of Future Science, Jilin University, Changchun 130012, P. R. China
First published on 29th January 2026
Abstract
Metal-doped ferrierite (FER) zeolites demonstrate excellent catalytic potential for the direct conversion of methane to methanol. In this study, density functional theory (DFT) calculations were employed to systematically investigate the reaction pathways and catalytic performance of active sites in metal-doped FER zeolites (M = Fe, Co, Ni, Cu). Theoretical analysis reveals that the C–H bond activation mechanism strongly depends on the electronic structure of the central metal, and metals with partially filled d-orbitals (Fe, Co, and Ni) tend to follow a concerted proton-electron transfer (CPET) pathway, whereas copper (Cu) with a fully occupied d10 configuration favors a hydrogen atom transfer (HAT) mechanism. Notably, the [CuO]+ active site, owing to its distinct electronic structure, exhibits both strong oxygen radical character and remarkable electron-withdrawing capability, leading to the highest reactivity in methane C–H bond activation. This work elucidates the structure–activity relationships of metal-doped zeolite active sites at the atomic and electronic levels, provides key theoretical insights into the core reaction mechanism, and offers valuable guidance for designing a new generation of highly efficient zeolite-based catalysts for the selective oxidation of methane to methanol.
1 Introduction
Methane, as a major component of abundant natural gas, holds significant potential for direct and selective conversion into high-value liquid fuels and chemicals (such as methanol), which remains one of the “Holy Grail” challenges in catalytic chemistry.1–4 Compared to current high-energy indirect conversion routes, the direct partial oxidation of methane to methanol under mild conditions offers substantial economic and environmental advantages. Among various catalysts, multi-phase catalysts represented by copper-exchanged zeolites (such as Cu-MOR4,5 and Cu-FER6) have demonstrated unique activity in achieving this transformation, particularly in aqueous reaction media or high-temperature gaseous oxygen conditions. Over the past two decades, research efforts have shifted from initially focusing on catalyst synthesis and performance optimization to gaining deeper insights into catalytic mechanisms, especially the critical C–H bond activation step, with a particular emphasis on understanding the pathways of electron and proton transfer.Early studies established the crucial role of copper-exchanged zeolites in methane conversion. Groothaert et al.7 firstly reported that Cu-ZSM-5 reacts with CH4 and O2 at high temperatures (>200 °C) to generate methoxy species, which subsequently hydrolyze to form methanol, revealing the potential core role of di-copper oxide species. Subsequent work, particularly through advanced techniques such as X-ray absorption spectroscopy (XAS), infrared spectroscopy (IR), and density functional theory (DFT) calculations, confirmed the nature of reactive oxygen species. For example, research by Alayon et al. 8 and Sushkevich et al. 9 demonstrated that in Cu-MFI, the active center is [Cu3(µ-O)3]2+, which can efficiently activate methane's C–H bonds. This discovery shifted the research focus to understanding the specific chemical mechanism of this activation step.
In C–H bond activation, proton-coupled electron transfer (PCET) plays a central role. PCET is a broadly theoretical concept, generally referring to any chemical reaction that involves both proton and electron transfer. Protons and electrons can originate from different molecular orbitals, and the transfer steps can be carried out simultaneously. The process most often proceeds via hydrogen atom transfer (HAT), in which a metal–oxo site abstracts a hydrogen atom (proton + electron) from methane, forming a methyl radical and a hydroxylated catalyst site.10 The transition state exhibits obvious radical characteristics. Its rate is highly correlated with bond dissociation energy (BDE), and the kinetic isotope effect (KIE) value is usually significant and is less affected by the polarity of the solvent. Alternatively, some systems may follow a concerted proton–electron transfer (CPET) pathway, where the proton and electron are transferred simultaneously but to different acceptors11,12 (Scheme 1). Experimentally, it is manifested as a non-linear dependence of the reaction rate on the driving force. The CPET mechanism requires the catalyst to possess dual functional characteristics of both proton acceptor and electron acceptor, and the two must be moderately coupled in terms of space and energy. For the C–H bond functionalization dominated by HAT, the design focus of the catalyst lies in regulating the radical properties of the M—O center to optimize the hydrogen atom affinity.
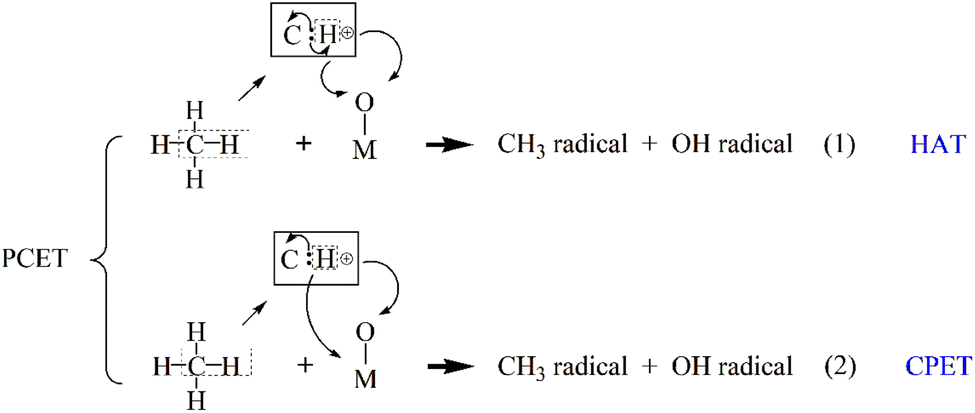 | ||
| Scheme 1 The classification of PCET mechanisms. | ||
Snyder et al.13 systematically analyzed metal-oxide-mediated C–H bond activation and clarified that many reactions previously classified under HAT are more accurately described by CPET in polar environments. The confined environment of zeolites stabilizes charged intermediates and facilitates proton migration to framework oxygen atoms, allowing the transfer pathways of electrons and protons to be spatially separated.
This theoretical development has been supported by specific studies on copper-zeolite systems. For example, Pappas et al. 14 combined transient dynamics, spectroscopy, and DFT calculations in their study of the Cu-MOR system, demonstrating that the methane activation step aligns more closely with the characteristics of PCET. In this process, proton transfer to the zeolitic framework oxygen acts as the rate-determining step, while electrons reduce the copper center. Additionally, Borfecchia et al.15 observed the simultaneous generation of zeolitic framework hydroxyl groups and the reduction of copper centers during methane activation, providing critical experimental evidence for the PCET mechanism. Recent studies have indicated that the distribution of products governed by HAT and CPET mechanisms is influenced not only by the composition and doping of oxide clusters but also by the electronic effects of ligands on metal centers.16 Akhil et al. revealed the decisive role of the electronic structure and spin state of the iron–oxygen core in the reaction mechanism.17 Therefore, in the field of zeolites, discussions on the two mechanisms and the precise relationship between dopant type, electron transfer dynamics, and catalytic activity remain insufficiently understood.
To address this gap, this study delves into constructing active sites in metal-doped FER zeolites and analyzing the corresponding reaction mechanisms. First, Fe2+, Co2+, and Ni2+ ions are introduced into FER motivated by their high magnetic properties to form bimetallic [Cu(µ-O)M]2+ (M = Fe, Co, Ni) active sites. The structural and energetic variations during C–H bond activation, methanol precursor formation, and methanol desorption were analyzed. Second, attention was directed to copper-only doping, including [CuO]+ [Cu2(µ-O)]2+, and [Cu3(µ-O)3]2+. These species, anchored on the FER framework, function as distinct active sites and can be represented as [Cux(µ-O)y]n+ clusters, where x denotes the number of copper centers, y the number of bridging oxygen atoms, and n the net charge of the species. For the [CuO]+, the oxygen atom is considered terminal rather than bridging. Through comparing these different metal loading configurations, we explored the structure–property relationships among structural characteristics, electronic properties, and methane activation performance. We particularly pay attention to the electron transfer mechanism during the catalytic process at different active sites, as well as the relationship between electron transfer and catalytic activity. Our goal is to develop high-activity, high-stability catalytic materials for direct methane-to-methanol conversion.
2 Computational details
All density functional theory (DFT) calculations in this study were performed using the Vienna Ab initio Simulation Package (VASP).18 The ion–electron interactions were described by the projector augmented wave (PAW) method, and exchange-correlation effects were described by the Perdew–Burke–Ernzerhof (PBE) functional19–21 based on the generalized gradient approximation (GGA). A plane-wave basis set was employed, with an energy cutoff of 550 eV. During structural optimization, ionic relaxation was carried out using the conjugate gradient method. To ensure the accuracy and reliability of the calculation results, a k-point convergence test was systematically conducted by gradually increasing the k-point density until the total energy changed by less than 1 meV per atom and only the Gamma point was used for k-point sampling. Under fully relaxed conditions for both shape and volume of the unit cell, we optimized a periodic topological model of pure silicon FER with the unit cell expanded once along the c-axis. The resulting lattice parameters are a = 19.118 Å, b = 14.321 Å, and c = 15.121 Å. All subsequent calculations were performed using a periodic model with fixed shape and volume. To compensate for the positive charge of the Cu complex on the framework, different Al3+ ions were introduced into the FER framework. For [Cu(µ-O)M]2+ [Cu2(µ-O)]2+ and [Cu3(µ-O)3]2+, two Al3+ ions were substituted for Si4+. For [CuO]+, only one Al3+ ion was substituted for Si4+. Structural optimization was converged when the forces on all atoms were less than 0.03 eV Å−1 and the change in electronic self-consistent field energy was less than 10−5 eV. The van der Waals correction (DFT-D3) proposed by Grimme22 was added to the energies obtained from the exchange correlation functional in all calculations to compensate for the long-range van der Waals dispersion interaction. Transition state searches were conducted using the climbing image nudged elastic band (CI-NEB) method combined with the improved dimer method (IDM).23,24 The Gibbs free energy was calculated based on frequency calculations using a VASPKIT software package.25 All Gibbs free energy corrections were calculated at 473 K, which corresponds to the temperature reported in the literature for zeolite-catalyzed methane-to-methanol conversion.4 During both structural optimization and transition state searches, all atoms were allowed to fully relax. The atomic charge densities were analyzed using Bader charge analysis program,26 atomistic density of states analysis and the combination of multiple peaks were analyzed using the VASPKIT program27 and the HSE06 hybrid functional calculations28 were employed to determine the charges at various sites (as shown in the SI).3 Results and discussion
This study employs DFT calculations to investigate the structure–activity relationships of [Cu(µ-O)M]2+ and [Cux(µ-O)y]n+ active sites in the direct conversion of methane to methanol. By comparing the activation free energy barriers of different active sites, a clear correlation between the active site configuration and catalytic performance was established. Additionally, the electronic transfer mechanism during C–H bond cleavage was elucidated through projected density of states (PDOS) analysis, while the relationship between electronic structure parameters and catalytic activity was further explored by examining the spin and charge densities of µ-O atoms.3.1 [Cu(µ-O)M]2+-FER
The optimized bimetallic [Cu(µ-O)M]2+-FER (M = Fe, Co, Ni) active sites at different spin multiplicities are shown in Fig. S1. DFT calculations indicated that two Al atoms preferentially occupy the T4 positions within the eight-membered ring, thereby providing a suitable coordination environment for the formation of subsequent active sites.As shown in Fig. 1a), during the catalytic conversion of methane over [Cu(µ-O)Fe]2+-FER, the quartet spin state exhibited a methane adsorption free energy (ΔGads) of −0.26 eV and a corresponding C–H bond breaking transition-state free energy barrier (ΔGa) of 1.10 eV. This activity was similar to that of the sextet state (ΔGads = −0.27 eV, ΔGa = 1.17 eV). Spin flips occurred before and after the adsorption process. Spin-polarized electron transfer pathway analysis reveals that, during C–H bond dissociation, β-spin electrons are localized on the carbon atom of the CH3 radical, while α-spin electrons participate in delocalized π* orbital hybridization through the 3d orbitals of Fe2+ and the Cu–O–Fe bonds. Additionally, when the quartet state intermediate forms, the carbon atom exhibits significant pronounced negative spin population (ρC = −0.432, see Table 1), and the spin population at the Fe center increases from 2.715 e to 3.414 e, confirming that Fe acts as the primary α-spin electron acceptor driving C–H bond cleavage. The energy change of the Fe 3d orbitals revealed the primary driving force behind this reaction: in the quadruply bonded Fe–O system, the α orbitals of Fe 3d initially contained four α electrons. Upon accepting an additional α electron from the CH4 1s/2p orbital, these five α electrons filled the Fe 3d α orbitals completely, resulting in a strong stabilizing effect and significantly lowering their energy (the peak shifted from −1.97 eV to around −2.67 eV). Thus, the C–H bond breaking process in methane catalyzed by [Cu(µ-O)Fe]2+-FER conforms to the typical CPET mechanism and serves as a contrast to the activation mode reported for Fe-ZSM-5 systems.29 The most notable feature of this process was the transfer of an α electron from the CH4 σ orbital to the FeO α orbitals, which filled the Fe 3d α orbitals and provided additional stabilization.
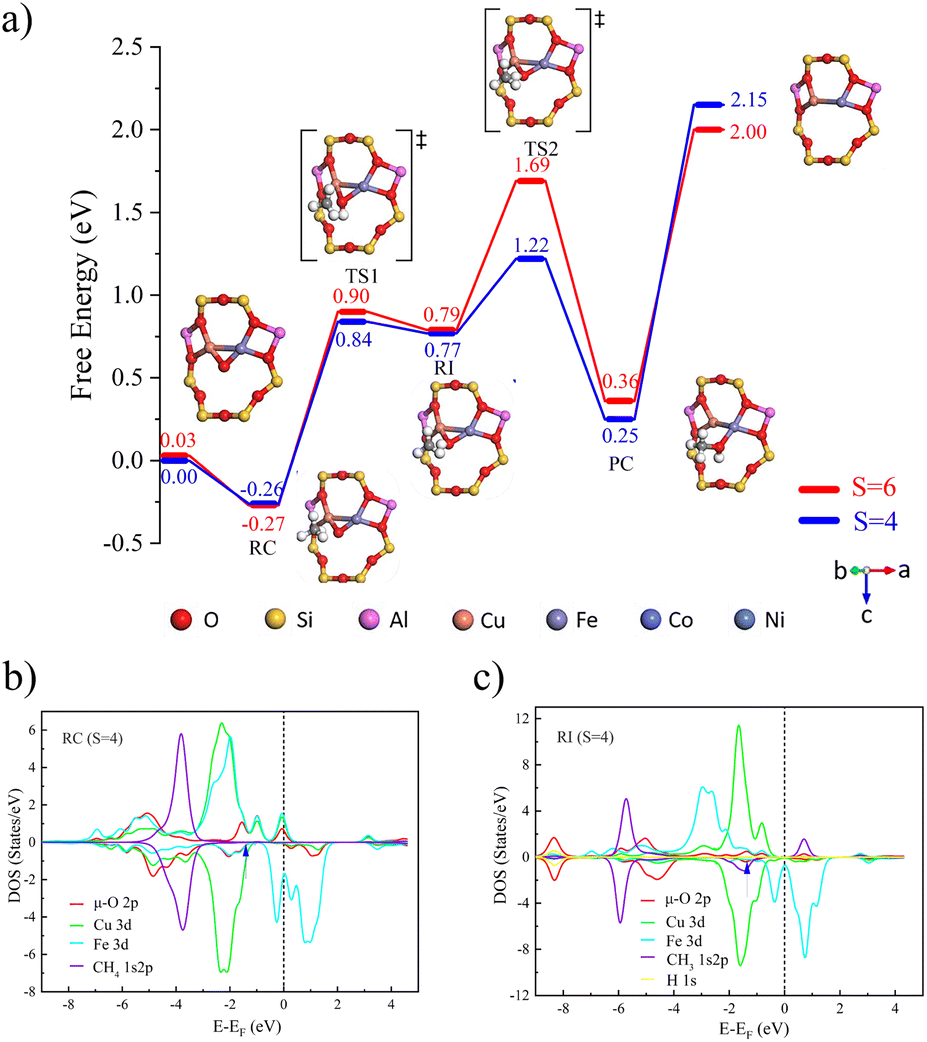 | ||
| Fig. 1 (a) Free energy diagram of [Cu(µ-O)Fe]2+-FER catalyzing CH4 to CH3OH at different multiplicities (red represents S = 6, blue represents S = 4). The PDOS of (b) the reactant complex and (c) radical intermediate (RI) catalyzed by [Cu(µ-O)Fe]2+-FER. The d-band centers are pointed by arrows and listed in Table S5. | ||
| Spin multiplicity | ρ(Cu) | ρ(Fe) | ρ(O) | ρ(C) | ρ(H) | |
|---|---|---|---|---|---|---|
| Reactant | 4 | −0.138 | 2.715 | 0.158 | 0.002 | 0.000 |
| Complex | 6 | 0.372 | 3.643 | 0.411 | 0.002 | 0.000 |
| Radical intermediate | 4 | 0.153 | 3.414 | 0.031 | 0.432 | 0.007 |
| 6 | 0.174 | 3.438 | 0.150 | 0.439 | 0.017 |
For Co- and Ni-doped FER zeolite catalysts of methane-to-methanol reactions, we observed that there is no M−O bond α spin 3d orbital about to be filled as in the [Cu(µ-O)Fe]2+ active site. When the C–H bond of methane breaks, the β electron on the CH4 1s2p σ orbital is transferred while the α electron remains on the C atom, causing the spin density of the C atom to increase from 0.00 to 0.428 (see Table S4). Therefore, [Cu(µ-O)Ni]2+ and [Cu(µ-O)Co]2+ react with methane via the HAT mechanism as well. Notably, in the case of [Cu(µ-O)Ni]2+-FER catalysis, the free energy barriers for the transition states in the doublet and quartet states were calculated to be 1.18 eV and 0.91 eV, respectively, which are lower than those observed for [Cu(µ-O)Co]2+-FER (1.17 eV, Fig. S2). Due to its higher catalytic activity, we focus our detailed discussion on the [Cu(µ-O)Ni]2+ system.
For [Cu(µ-O)Ni]2+-FER, the difference in spin population for the doublet intermediate reveals a decrease of 0.693 at the Ni center, confirming that Ni2+ serves as the primary acceptor for β-spin electrons (Table S4). Based on the description above, in the process of methane catalysis by [Cu(µ-O)Ni]2+-FER, due to the electronic deficiency in the valence orbitals of Ni, electrons tend to transfer towards Ni during hydrogen transfer, while protons are accepted by oxygen. According to the exchange-enhanced reactivity (EER)30 principle, for a doublet structure with one unpaired α electron, the transfer of β electrons from the donor orbital (1s-2p orbital of methane) to the acceptor orbital (Cu–O–Ni π* antibonding orbital) is unfavorable.31 However, in this reaction, the β electrons are transferred to the Ni 3d-β orbital, which has a favorable exchange interaction with the existing unpaired Ni 3d-α orbital. This exchange interaction stabilizes the orbital energy and reduces electronic repulsion in the acceptor orbital, leading to higher catalytic activity of [Cu(µ-O)Ni]2+ compared to [Cu(µ-O)Co]2+. Ultimately, the catalytic activity order for [Cu(µ-O)M]2+ type active sites is as follows [Cu(µ-O)Ni]2+ > [Cu(µ-O)Fe]2+ > [Cu(µ-O)Co]2+.
By calculating the spin population and Bader charge distribution of each active site, the correlation between the electronic structure parameters and catalytic performance in the [Cu(µ-O)M]2+ type active sites was established. The charge densities of the bridged oxygen atoms decrease in the order [Cu(µ-O)Fe]2+ > [Cu(µ-O)Co]2+ > [Cu(µ-O)Ni]2+, whereas they increase in the same sequence (Table S6). This inverse relationship indicates that active sites with higher spin populations generally possess less negative charge on the µ-O atom. Consistent with previous studies, bridging oxygen atoms with lower negative charge and higher spin polarization are more favorable for promoting C–H bond cleavage in methane.32
To analyse the reasons for the differences in the catalytic activity of [Cu(µ-O)M]n+ type active sites, we investigated the electronic structures of the µ-O 2p orbitals. First, in [Cu(µ-O)Fe]2+, the 2px orbital of O crossing the Fermi level was analyzed; due to its strong hybridization with the 3d orbitals of Cu and Fe atoms outside the framework, a delocalized d-p π bond was formed (see Fig. S3). Its conjugation effect helps break the C–H bond via the CPET mechanism. However, differences in the C–H bond breaking mechanisms prevent [Cu(µ-O)Fe]2+ from achieving the high catalytic efficiency observed in biological iron–oxo complexes.
The significant electronic spin mismatch between the µ-O 2py orbital and the α-HOMO and β-LUMO, the energy-lowering effect of electron exchange interactions on the system is severely weakened, resulting in lower catalytic activity for [Cu(µ-O)Co]2+. The PDOS plots of µ-O for [Cu(µ-O)Ni]2+ and [Cu(µ-O)Co]2+ are similar in overall shape (see Fig S4 and S5), but the β-LUMO energies of the µ-O 2px, 2py, and 2pz orbitals in [Cu(µ-O)Ni]2+ are all lower than those of the corresponding orbitals in [Cu(µ-O)Co]2+, leading to higher catalytic activity.
3.2 [Cux(µ-O)y]n+-FER
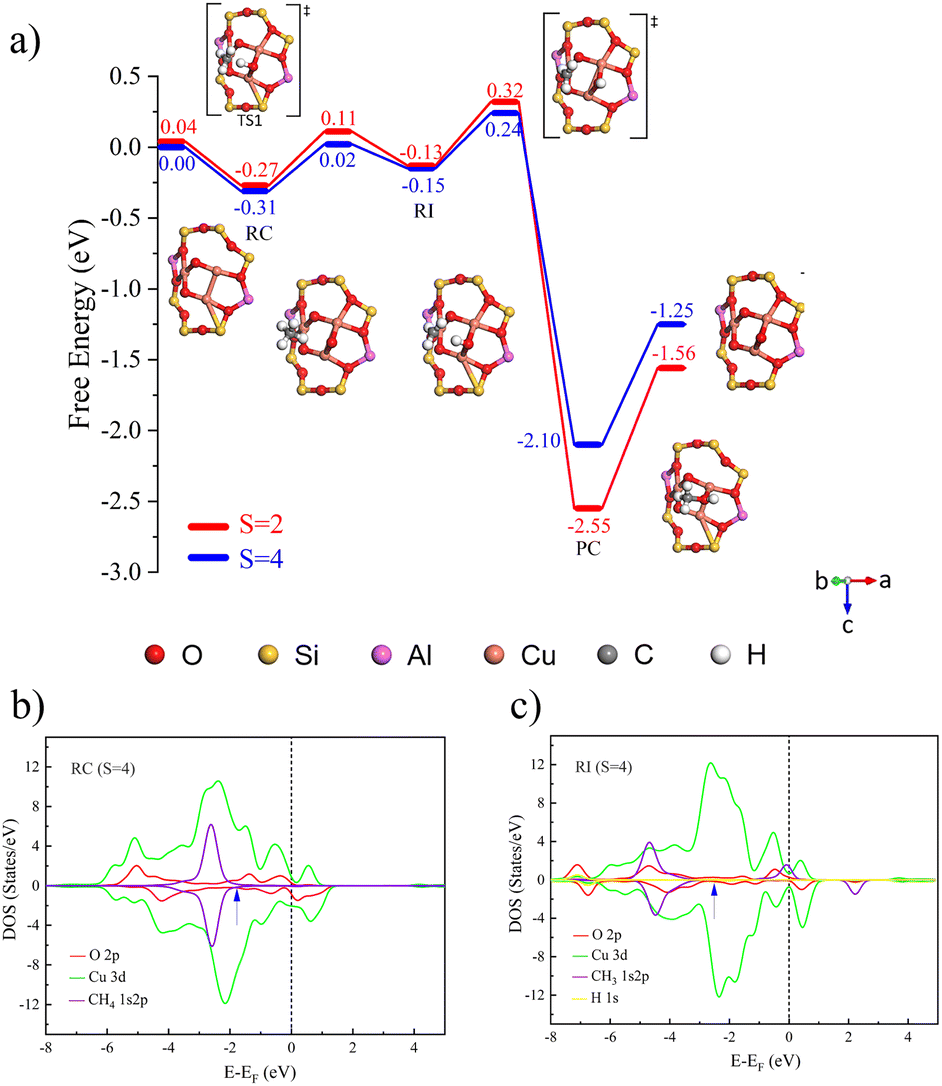
Having understood the fundamental characteristics of [Cu(µ-O)M]2+-FER systems, we further expanded our research scope to explore [Cux(µ-O)y]n+-FER systems with varying compositions. By tuning the values of x and y, these diverse structures exhibited a wider range of properties and potential applications.In this section, we focus on three copper-oxo active center models with typical structural features, mononuclear [CuO]+, binuclear [Cu2(µ-O)]2+, and trinuclear [Cu3(µ-O)3]2+ systems. Fig. S6 shows the structure of the [Cux(µ-O)y]n+-FER and both Al atoms are located at the T4 positions of the eight-membered ring. For the [Cux(µ-O)y]n+-FER systems under different spin populations, we performed comprehensive structural optimization and obtained the most stable configurations of each active site under different spin multiplicities. To evaluate the thermodynamic stability of the active sites, this work referred to the research of Wang et al.,33 and calculated the formation energy of each site under different spin multiplicities with the Cu-FER as the benchmark (see Table S8). All the oxygen-active species at the active sites in this article were generated by oxygen excitation. The results show that the formation of all active sites is thermodynamically favorable, and the stability is arranged in the order [Cu3(µ-O)3]2+ > [Cu2(µ-O)]2+ > [CuO]+. Taking [Cu3(µ-O)3]2+ as an example (the numbering of Cu and O atoms is detailed in Fig. S7), our calculations reveal that the µ-O2 and µ-O3 atoms exhibit equivalent chemical environments. Based on this, in subsequent studies, we primarily focused on the µ-O1 and µ-O2 sites to deeply explore their catalytic performance.
The process of catalyzing methane conversion to methanol begins with the formation of a precursor complex. During the reaction process, the methane C–H bond is activated by adsorption onto the catalytically active site. Specifically, the C–H bond of methane is cleaved after overcoming the first transition state, and the hydrogen radical extracted combines with the µ-O at the active site, forming a bridged hydroxyl radical, while a methyl radical is also generated. These two radicals together constitute the key intermediate species (Fig. 2a)). Following the first transition state, the system undergoes a spin-crossover reaction.34 This phenomenon manifests as an intersection between potential energy surfaces of different spin multiplicities, the singlet-state structure gradually gains energetic dominance, and the process of converting from triplet to singlet states is defined as spin crossover. The realization of this process is attributed to the synergistic effects of tunnelling and spin inversion probability (SIP), which effectively lower the activation energy barrier.35
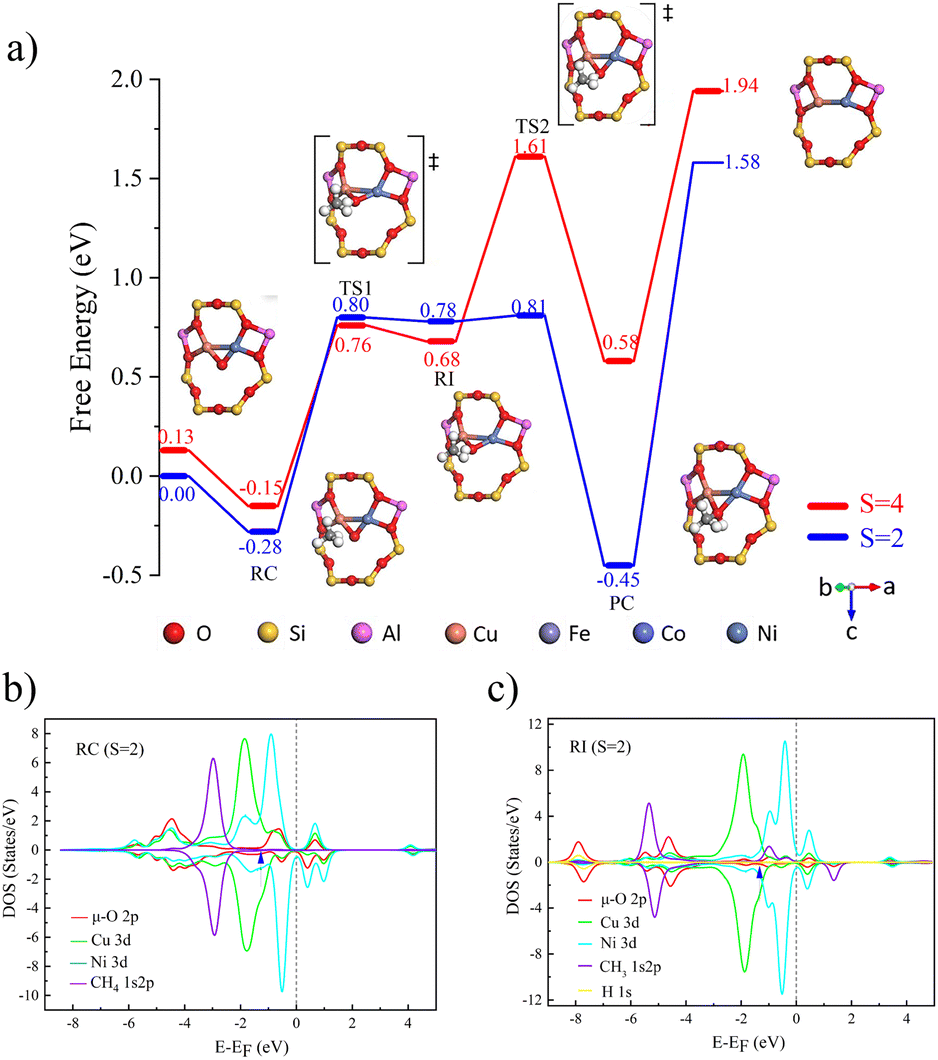 | ||
| Fig. 2 (a) Free energy diagram of [Cu(µ-O)Ni]2+-FER catalyzing CH4 to CH3OH at different multiplicities (red represents S = 4, blue represents S = 2). The PDOS of (b) the reactant complex and (c) radical intermediate (RI) catalyzed by [Cu(µ-O)Ni]2+-FER. The d-band centers are pointed by arrows and listed in Table S5. | ||
The activation of the C–H bond proceeds via a distinct electron transfer process wherein the β-electron from the σ orbital of methane is transferred into the π* antibonding orbital of the Cu–O moiety. This transfer is quantitatively supported by spin population analysis. The data demonstrate a marked decrease in the spin population on the µ-oxo ligand, from 1.003 to 0.375, accompanied by a reduction on the Cu centers from 0.605 to 0.511. This pronounced loss of spin density on the µ-O site directly indicates that it serves as the primary acceptor for the incoming β-electron. Conversely, the corresponding α-electron remains localized on the methane carbon atom, as evidenced by an increase in its spin population from 0.000 to 0.449 (Table 2), generating a proton and carbon-centered radical. Given the closed-shell d10 configuration of Cu, which precludes significant electron uptake at the metal center, both the proton and the electron pair are ultimately accepted by the oxygen atom. The concerted yet asynchronous transfer of a hydrogen atom exclusively to the oxygen center confirms that the C–H bond cleavage in [CuO]+ follows the HAT mechanism.
| Spin multiplicity | ρ(Cu) | ρ(O) | ρ(C) | ρ(H) | |
|---|---|---|---|---|---|
| Reactant | 1 | −0.073 | 0.082 | 0.000 | 0.000 |
| Complex | 3 | 0.605 | 1.003 | 0.000 | 0.000 |
| Radical intermediate | 1 | 0.005 | 0.001 | −0.003 | 0.000 |
| 3 | 0.511 | 0.375 | 0.449 | 0.007 |
The C–H bond of methane undergoes cleavage after experiencing a transition state, with free energy barriers for singlet and triplet states being 0.18 and 0.21 eV, respectively. As shown in Fig. 3b and c), because the energies and peak shapes of Cu atoms and µ-O atoms at the Cu–O π* anti-bond orbitals are very close, we can indicate that a strong bond is formed between Cu and µ-O after the transition state. By analogy with [CuO]+ [Cu2(µ-O)]2+ forms singlet and triplet complexes with a binding free energy of 0.29 eV. In the catalytic process of methane conversion, this system also cleaves C–H bonds via the HAT mechanism, with free energy barriers for the two transition states being 0.50 and 0.54 eV (Fig. S8a)).
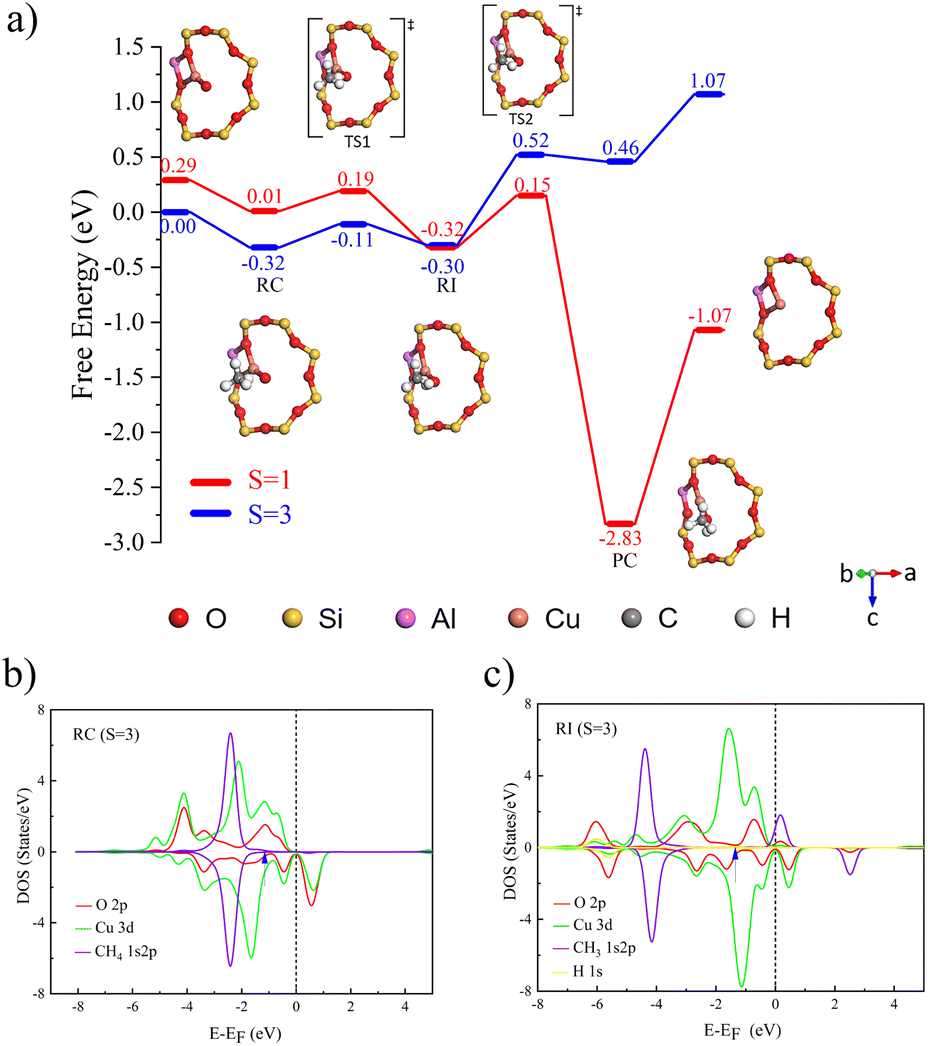 | ||
| Fig. 3 (a) Free energy diagram of [CuO]+-FER catalyzing CH4 to CH3OH at different multiplicities (red represents S = 1, blue represents S = 3). The PDOS of (b) the reactant complex and (c) radical intermediate (RI) catalyzed by [CuO]+-FER. The d-band centers are pointed by arrows and listed in Table S15. | ||
Following the C–H bond cleavage in [CuO]+, the reaction proceeds through a second singlet-state transition state (free energy barrier of 0.47 eV), leading to the formation of a stable product complex. It is important to note that while the calculated energy required for product detachment is relatively high, under actual reaction conditions with solvents present, this energy demand will be significantly reduced. Theoretical studies in the literature have shown that introducing solvent molecules, such as water, can typically reduce the energy required for product detachment to less than half of its original value.36
Further investigation was conducted on the reaction mechanism of methane selective oxidation to methanol catalyzed by [Cu3(µ-O)3]2+. By comparing the catalytic performance differences between the µ-O1 and µ-O2 sites, it was found that although both sites follow the HAT mechanism for C–H bond activation, their catalytic activities exhibit significant differences. In the reaction pathway mediated by µ-O1, the free energy barriers for the two transition states are 0.16 eV and 0.34 eV (Fig. 4a)). It can be referred that the β electrons from the σ orbitals of methane are primarily transferred to the Cu–O–Cu π* antibonding orbital. Orbital composition analysis shows that this β-LUMO acceptor orbital (at an energy level of 0.54 eV) is predominantly dominated by the 2p orbitals of the oxygen radical, with a relatively low contribution from the 3d orbitals of copper (Fig. 4b and c). Notably, in the quartet reaction center structure, the spin population at the µ-O1 site reaches as high as 0.73, confirming its significant oxygen radical characteristics.
 | ||
| Fig. 4 (a) Free energy diagram of [Cu3(µ-O)3]2+-FER (µ-O1) catalyzing CH4 to CH3OH at different multiplicities (red represents S = 2, blue represents S = 4). The PDOS of (b) the reactant complex and (c) radical intermediate (RI) catalyzed by [Cu3(µ-O)3]2+-FER (µ-O1). The d-band centers are pointed by arrows and listed in Table S15. | ||
In contrast, the orbital hybridization features of the µ-O2 site exhibit fundamental differences. As shown in Fig. S9, the contribution of the 3d orbitals of Cu in its π hybridized orbitals is increased, leading to a significant weakening of the oxygen radical characteristics (with a spin population of only 0.21). This difference in the electronic structure is directly reflected in the comparison of transition state energy barriers by comparing the activation energies of the first transition state for C–H bond cleavage across various catalytic systems, and the order of catalytic activity was determined as follows [CuO]+ > [Cu3(µ-O)3]2+(µ-O1) > [Cu2(µ-O)]2+ > [Cu3(µ-O)3]2+(µ-O2). The conclusions drawn in this work regarding the catalytic ability sequence of each copper active site are basically consistent with those of Wang et al.33 On this basis, a discussion on the catalytic ability of different µ-O atoms in [Cu3(µ-O)3]2+ was added. This energy barrier difference is negatively correlated with the radical characteristics of the oxygen sites, confirming that the µ-O1 site, with its strong radical properties, is more favorable for activating the C–H bonds via a single-electron transfer mechanism. Consequently, it exhibits superior catalytic performance.
Taking [CuO]+ and [Cu2(µ-O)]2+ sites as the main research objects, we found that although traditional surface catalysis theory suggests that µ-O sites of binuclear sites have superior catalytic performance, mononuclear sites exhibit unique electronic structure advantages in methane C–H bond activation. The β-LUMOs of the triplet [CuO]+ site show significant density peaks at 0.45 eV (2py orbital) and 0.73 eV (2pz orbital), with peak intensities reaching 3.8 eV and 4.2 eV, respectively (Fig S10). These high-density acceptor orbitals indicate that the mononuclear site has excellent electron-capturing ability, effectively promoting the transfer of β-electrons from the σ-orbital of methane to the Cu–O π* anti-bonding orbital, thereby efficiently activating C–H bonds via a HAT mechanism, resulting in the higher catalytic capacity.
In contrast, the β-LUMOs of [Cu2(µ-O)]2+ sites show only weak density peaks at 0.54 and 0.48 eV (Fig. S11), with significantly reduced electron-capturing ability. This electronic structure characteristic leads to a decrease in the efficiency of β-electron transfer from methane to Cu–O–Cu π* orbitals, thereby weakening the catalytic activity of the binuclear system. Notably, the excellent performance of mononuclear sites is closely related to their unique electronic configuration, and the confinement effect provided by the pore structure of the zeolite not only stabilizes the [CuO]+ structure but also regulates the electronic distribution of Cu–O bonds, allowing the 2p orbitals of oxygen to dominate in the β-LUMO. This highly localized acceptor orbital property significantly enhances the single-electron transfer capability of mononuclear sites, achieving efficient homolytic cleavage of C–H bonds.
We explore the mechanisms by which different active sites catalyze C–H bond breaking. By comparing [Cu(µ-O)M]n+ and [Cux(µ-O)y]n+ active sites, our study reveals distinct mechanistic preferences based on the electronic state of the central metal atom. Specifically, we find that active sites with an electron-deficient central metal tend to follow a CPET mechanism. Their electrons and protons are transferred to different receptors. In contrast, when the central metal is in a closed-shell configuration with fully filled d orbitals, the ability of electrons to transition in the system is relatively weak, and they tend to transfer together with protons as a whole. Then, the process leans toward a HAT mechanism. In biological systems, where active metal centers are typically Fe, Co, or Ni, CPET mechanisms dominate due to the electronic structure of these metals. However, within the confined environment of zeolite channels, the charge density on active oxygen species becomes a critical factor in determining the reaction pathway. This is particularly evident in mono-copper sites, where copper ions possess fully filled d10 configurations. This electronic feature enhances their ability to provide feedback π electrons to antibonding π* orbitals µ-O, making it challenging for the charge density on the bridge oxygen to increase significantly. This structural property ultimately dictates that mono-copper active sites primarily facilitate C–H bond cleavage through HAT mechanisms rather than CPET. The energy matching of receptor orbital levels plays a crucial role in controlling catalytic activity, as evidenced by the comparable transition state free energy barriers observed during methane activation for both trinuclear and binuclear sites (as shown in Fig. S11–13).
To establish a correlation between electronic structural parameters and catalytic performance, we systematically calculated the spin population and Bader charge distribution for each active site (Table S13). It revealed that the O at the [CuO]+ site exhibits the highest spin population, corresponding to the strongest oxygen radical character. The µ-O1 in the tri-copper site follows, while the µ-O in the binuclear site and the µ-O2 in the tri-copper site show significantly weakened radical activity. Further analysis found that the spin population of µ-O is strongly negatively correlated with its net charge. This discovery aligns closely with previously reported trends; µ-O sites in zeolite confined environments with high spin population and low negative charge can significantly lower the C–H bond activation free energy barrier by enhancing single-electron transfer capability.32 Additionally, we compared the spin populations of two types of active sites, the µ-O in heterometallic [Cu(µ-O)M]2+ sites generally exhibits lower overall spin population. From this, it can be inferred that [Cu(µ-O)M]2+ sites are more energetically stable, but their ability to catalyze C–H bond cleavage may be somewhat diminished, consistent with existing studies that describe a negative correlation between the stability of active sites and their methane activation activity.37
4 Conclusions
This study provides valuable theoretical insights into the catalytic performance of various active sites in metal-doped FER zeolites for methane selective oxidation to methanol. By comparing the free energy barriers of different active sites, we establish the following catalytic activity order; for the [Cu(µ-O)M]2+ type active sites (M = Fe, Ni, Co), [Cu(µ-O)Ni]2+ shows the best catalytic ability, and, for the [Cux(µ-O)y]n+ type active sites, [CuO]+ is the active site with the strongest catalytic ability. Our findings reveal that the C–H bond cleavage mechanism varies significantly with the electronic state of the central metal: electron-deficient metals favor a concerted proton–electron transfer (CPET) pathway, whereas copper with a fully occupied d10 configuration prefers hydrogen atom transfer (HAT). In addition, our result suggests that the [CuO]+ site demonstrated superior catalytic performance due to its strong oxygen radical character and efficient electron attracting capability. This work highlights the critical role of the actual hybridization state of transition metal d orbitals in analyzing heterometallic active sites and offers important references for advancing research on transition metal-doped zeolite catalytic reactions. By filling the theoretical gap of the proton-electron transfer mechanism of metal-doped FER active sites in methane–methanol conversion, this study provides essential guidance for the development of new zeolite catalysts with enhanced performance for efficient methane-to-methanol conversion.Author contributions
Zirong Guan: writing – original draft, visualization, investigation, formal analysis, data curation. Jiaming Miao: writing – original draft, investigation, formal analysis, data curation. Wenfu Yan: project administration. Zexing Qu: writing – review & editing, supervision, methodology, project administration Yi Li: writing – review & editing, supervision, resources, project administration.Conflicts of interest
There are no conflicts to declare.Data availability
Most of the data supporting this article have been included as part of the supplementary information (SI) document, and additional data are available from the corresponding author upon reasonable request. Supplementary information is available. See DOI: https://doi.org/10.1039/d5ta10400a.Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (No. 22175073, 22373040, and 22288101) and Science & Technology Development Program of Jilin Province (No. 20230101349JC). We also gratefully acknowledge the High Performance Computing Center of Jilin University for providing computation support.References
- M. N. Mu-Hyun Baik and R. A. Friesner, Chem. Rev., 2003, 103, 2385–2419 CrossRef PubMed.
- R. Sharma, H. Poelman, G. B. Marin and V. V. Galvita, Catalysts, 2020, 10(2), 194–219 CrossRef CAS.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 1835–1844 CrossRef CAS.
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Román-Leshkov, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed.
- M. B. Park, S. H. Ahn, A. Mansouri, M. Ranocchiari and J. A. van Bokhoven, ChemCatChem, 2017, 9, 3705–3713 CrossRef CAS.
- M. S. Kim, K. H. Park, S. J. Cho and E. D. Park, Catal. Today, 2021, 376, 113–118 CrossRef CAS.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, ACS Catal., 2018, 8, 8306–8313 CrossRef CAS.
- D. P. V. L. Sushkevich, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef PubMed.
- M. L. Neidig, C. D. Brown, M. Kavana, O. W. Choroba, J. B. Spencer, G. R. Moran and E. I. Solomon, J. Inorg. Biochem., 2006, 100, 2108–2116 CrossRef CAS PubMed.
- C. T. Saouma, W. D. Morris, J. W. Darcy and J. M. Mayer, Chem. Eur J., 2015, 21, 9256–9260 Search PubMed.
- J. Li, S. Zhou, J. Zhang, M. Schlangen, T. Weiske, D. Usharani, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 2016, 138, 7973–7981 CrossRef CAS PubMed.
- B. F. Sels and B. E. R. Snyder, Science, 2017, 373, 327–331 Search PubMed.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- L. Yue, J. Li, S. Zhou, X. Sun, M. Schlangen, S. Shaik and H. Schwarz, Angew. Chem., Int. Ed., 2017, 56, 10219–10223 CrossRef CAS PubMed.
- A. Bhardwaj and B. Mondal, Inorg. Chem., 2025, 64, 14999–15010 CrossRef CAS PubMed.
- J. F. G. Kresse, Comput. Mater. Sci., 1996, 6, 15–50 Search PubMed.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 Search PubMed.
- D. J. G. Kresse, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- K. B. J. P. Perdew and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154123 CrossRef PubMed.
- G. Henkelman, D. Sheppard and R. Terrell, J. Chem. Phys., 2008, 128, 134106–134116 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- N. X. V. Wang, J. Liu, G. Tang, W. Geng, arXiv, 2019, preprint arXiv:1908.08269v08266, DOI:10.48550/arXiv.1908.08269.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033–108065 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 Search PubMed.
- C. B. Sun, M. W. Guo, S. S. Siwal and Q. B. Zhang, J. Catal., 2020, 381, 454–461 CrossRef CAS.
- D. Janardanan, Y. Wang, P. Schyman, L. Que and S. Shaik, Angew. Chem., Int. Ed., 2010, 49, 3342–3345 CrossRef CAS PubMed.
- A. Kazaryan and E. J. Baerends, ACS Catal., 2015, 5, 1475–1488 CrossRef CAS.
- Y. S. Kazunari Yoshizawa, T. Yumura and T. Yamabe, J. Chem. Phys., 2000, 104, 734–740 CrossRef.
- G. Wang, W. Chen, L. Huang, Z. Liu, X. Sun and A. Zheng, Catal. Today, 2019, 338, 108–116 CrossRef CAS.
- D. K. Hajime Hirao, L. Que Jr. and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590–8606 CrossRef PubMed.
- S. J. Klippenstein, V. S. Pande and D. G. Truhlar, J. Am. Chem. Soc., 2014, 136, 528–546 Search PubMed.
- M. H. Mahyuddin, T. Tanaka, Y. Shiota, A. Staykov and K. Yoshizawa, ACS Catal., 2018, 8, 1500–1509 Search PubMed.
- A. R. Kulkarni, Z.-J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Catal. Sci. Technol., 2018, 8, 114–123 Search PubMed.
Footnote |
| † Zirong Guan and Jiaming Miao contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2026 |