DOI:
10.1039/D5TA10370C
(Paper)
J. Mater. Chem. A, 2026,
14, 16104-16113
Transient behavior of self-healable ultra-stretchable carboxylic acid-doped polyaniline films for sustainable and re-processable polymer electronics
Received
19th December 2025
, Accepted 5th March 2026
First published on 5th March 2026
Abstract
This work evaluates the degradation capabilities of transient carboxylic acid-based dopants (CABDs), focusing on 1,2,4-benzene tricarboxylic acid (BA), citric acid (CA), and diphenic acid (DA). The stretchable electronic polymer complex is composed of polyaniline (PANI), poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAAMPSA), and CABDs. The film is synthesized through the oxidative polymerization of aniline while PAAMPSA acts as a template to guide the PANI polymerization. Structural variations among the dopants—including acidity, aromaticity, and rigidity—significantly influenced conductivity, mechanical properties, water retention, and self-healing efficiency. The PANI/PAAMPSA/BA composite exhibited the highest conductivity (0.0063 S m−1), while PANI/PAAMPSA/CA demonstrated exceptional stretchability (elongation at break of 3823%), the greatest water retention (15.1%), and a complete conductivity self-healing efficiency (100%). The degradation tests were carried out under soil burial and aqueous conditions. Interestingly, the films completely dissolved in distilled water, tap water and river water within 10 minutes. In addition, the dissolved solution could be recast to develop new functional sensors, indicating the reusability of the sensors. Soil degradation tests further demonstrated the degradation of the film within 24 hours. These findings confirm the potential of carboxylic acid-doped polymeric sensors as sustainable, eco-friendly materials for sensing applications that combine efficient degradability with re-processibility to minimize environmental impact.
1 Introduction
The growing demand for flexible and stretchable wearable technologies in diverse fields such as biomedical monitoring,1 soft robotics,2,3 environmental sensing,4–7 and human–machine interfaces8,9 has increased concerns regarding their end-of-life environmental impact. Conventional strain sensors, often composed of non-biodegradable materials with single-use capability, exacerbate the sustainability and pollution challenges across the globe. This has spurred the need for sustainable electronic devices that combine both electrical and mechanical properties with end-of-life recyclability or re-processibility.10 The development of transient electronics-a class of devices designed to perform their function and then physically disintegrate after their operational lifetime presents a promising strategy to reduce the impact of non-biodegradable materials.11 The intrinsically conducting polymer polyaniline (PANI) has received great attention because of its tunable electrical conductivity, facile synthesis, environmental stability, and low cost.12–17 However, pristine PANI suffers from drawbacks such as limited stretchability, low solubility, and non-biodegradability, which restrict its application in wearable stretchable electronics.18–20 The rigid backbone structure of polyaniline makes it difficult to break down by natural processes and raises significant concerns about long-term sustainability and environmental safety.21 Addressing the non-biodegradable nature of PANI is crucial for the development of eco-friendly sensor technologies. Researchers have incorporated biodegradable materials-e.g., cellulose,22,23 lignin,24,25 gelatin,26 chitosan27,28 – with PANI to reduce the environmental impact of non-biodegradable PANI-based systems.
Poly(2-acrylamido-2-methyl-1-propane sulfonic acid) (PAAMPSA) is a hydrophilic polymer electrolyte with a sulfonic acid and amide groups, which protonates PANI from the emeraldine base to its salt form.29–31 The ionizable sulfonic acid groups impart anionic character to PAAMPSA with excellent conductivity and mechanical properties.32 Regardless of these advantages, PAAMPSA's synthetic backbone and sulfonate groups makes it non-biodegradable. The polymer resists the microbial attack and environmental breakdown limits its biodegradability in natural conditions and can pose challenges for sustainable material development. PANI is often doped with PAAMPSA, which improves the aqueous processability and mechanical properties through ionic interactions.32 Additionally, PAAMPSA acts as a template that guides the in situ oxidative polymerization of aniline.29Doping of PANI/PAAMPSA with small molecule dopants often helps to tune the electrical and mechanical properties.33 In this work, carboxylic acid–based dopants (CABDs) are utilized, which contain one or more –COOH groups consisting of a carbonyl and a hydroxyl group bonded to the same carbon atom.34 The CABDs, including 1,2,4-benzenetricarboxylic acid, citric acid, and diphenic acid are investigated. These dopants were selected for their varied molecular structures, functional group arrangements, effect on protonation, intermolecular interactions, and environmental degradability. The structural differences among the dopants are expected to significantly influence the PANI/PAAMPSA-polymer network through variations in rigidity, aromaticity and hydrogen bonding potential. BA's rigid aromatic core promotes π–π stacking and ordered packing, CA's flexible and polar structure enhances hydrogen bonding and chain mobility, and DA's bulky biphenyl structure introduces steric hindrance that limits chain rearrangement. The –COOH groups in the carboxylic acid act as proton donors to the nitrogen sites along the PANI backbone. After the proton is accepted, the insulating emeraldine base form of PANI is converted to the conducting emeraldine salt form. Furthermore, carboxylate groups (–COO−) can form hydrogen bonds with both PANI and PAAMPSA, influencing the morphology and mechanical flexibility. Additionally, carboxylic acid groups impart degradability to the PANI/PAAMPSA composite films by increasing the hydrophilicity of the film.35 The hydrolytic nature causes the film to soften or swell by reducing the intermolecular forces between the polymer chains. This results in chain scission, fragmentation, and molecular weight reduction. The highly entangled network of polymer chains starts to slowly break down into smaller oligomers and monomers. Finally, smaller fragments caused by hydrolysis can be consumed by bacteria or fungi, leading to complete biodegradation of the original material.36–39 Compared to the stronger acids such as sulfonic and phosphoric acid, carboxylic acids offers a more environmentally friendly alternative for doping PANI. Many microorganisms possess enzymes that can recognize and break the carboxylated sites in the polymer.40–42 The combination of PANI, PAAMPSA and carboxylic acid yields a re-processable, conductive, and environmentally responsive composite film. In this work the in situ oxidative polymerization of CABDs with PANI/PAAMPSA resulted in a stretchable, flexible, conductive, and self-healable films. The structural differences among the dopants, particularly the differences in aromaticity, acidity, and rigidity, proved to strongly influence the electrical conductivity, mechanical properties, and self-healability. The sensors are found to be self-healable as well as re-processable. Degradation studies were performed in soil, tap water, distilled water, and river water. While numerous studies have reported degradable PANI-based systems, a systematic comparative analysis of how structurally distinct carboxylic acid dopants can be strategically tuned to balance conductivity, stretchability, and degradability remains largely unexplored. PANI/PAAMPSA/carboxylic acid doped composites hold strong potential for the development of transient electronics and sustainable sensing platforms, where controlled degradation and end-of-life recyclability are critical.
2 Results
2.1 Structure of dopants
The structure of carboxylic acid based dopants–1,2,4 benzenetricarboxylic acid (BA) (Fig. 1a), citric acid (CA) (Fig. 1b), and diphenic acid (DA) (Fig. 1c)–have a strong influence on the properties of the PANI/PAAMSA (Fig. S2) system. To elucidate these effects, the chosen dopants vary in molecular geometry, number of functional groups, steric hindrance, inter alia. This in turn will influence the hydrogen bonding potential, protonation efficiency, and overall morphology of the polymer complex.BA consists of three carboxylic acid groups on a single aromatic ring. The presence of multiple functional groups provide multiple protonation sites that can form electrostatic interactions and hydrogen bond with the polymer matrix. Additionally the aromatic ring allows for π–π stacking with the PANI chains, resulting in an orderly arrangement.43 CA also consists of three carboxylic acid groups, and is highly polar in nature making it water soluble.34 Multiple acid groups and the central hydroxyl groups allow it to form multiple hydrogen bonds with amine/imine groups of PANI and the amide and sulfonic acid groups of PAAMPSA. The small molecular size and flexible aliphatic backbone can result in an increased flexibility and stretchability of the doped film. DA, on the other hand, is composed of two benzene rings connected by a single bond with carboxylic acid groups located at the para positions. Among the three dopants investigated, DA exhibits the highest aromaticity and steric hindrance, due to its biphenyl linkage.44 This aromatic biphenyl core can align with the benzenoid and quinoid segments of PANI, and introduces π–π interactions. The two carboxylic acid groups can form hydrogen bonds with PANI and PAAMPSA, further influencing the structural organization and properties of the composites.
 |
| | Fig. 1 Structure of carboxylic acid based dopants (a) 1,2,4 benzenetricarboxylic acid (BA), (b) citric acid (CA), (c) diphenic acid (DA). | |
2.2 Infrared spectroscopy
The FTIR spectroscopy for the three PANI/PAAMPSA/CABDs is shown in Fig. 2. The bands at 1560 and 1500 cm−1 are assigned to the ring stretching vibration of the quinoid (Q) and benzenoid (B) units, respectively. The bands at 1296 and 1136 cm−1 correspond to the stretching vibration of C–N–C and B–NH+ = Q, respectively.45,46 The band at 1208 cm−1 is attributed to the asymmetric vibration of the sulfonic acid group (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). The absorption peak around 1640 cm−1 was attributed to the OH deformation mode of water. The peaks at 1032 cm−1 and 623 cm−1 are attributed to the SO3H and N–H (amide), respectively, originating from PAAMPSA.47 The CO stretching of the carboxylic acid appears at 1698 cm−1.48 For the PANI/PAAMPSA/DA film, this peak is not visible in the full spectrum; however, the enlarged region in Fig. S3 confirms the presence of the CO stretching vibration. The variation in the intensity, shape, and position of the CO stretching peak can be attributed to differences in hydrogen bonding, electrostatic interactions, and protonation effects when PANI/PAAMPSA interacts with the dopants. The control sample (Fig. S4) show a distinct peak at 1638 cm−1, which is assigned to the amide I CO vibrations of the PAAMPSA backbone49,50 shown in Fig. S5. A weak shoulder peak at 1723 cm−1 may be attributed to a minor fraction of free (non-hydrogen-bonded) carbonyl groups within the polymer matrix.51 The OH stretching band located at 3100–3600 cm−1 indicates hydrogen bonding between PANI, PAAMPSA, and the carboxylic-acid-based dopants.30
O). The absorption peak around 1640 cm−1 was attributed to the OH deformation mode of water. The peaks at 1032 cm−1 and 623 cm−1 are attributed to the SO3H and N–H (amide), respectively, originating from PAAMPSA.47 The CO stretching of the carboxylic acid appears at 1698 cm−1.48 For the PANI/PAAMPSA/DA film, this peak is not visible in the full spectrum; however, the enlarged region in Fig. S3 confirms the presence of the CO stretching vibration. The variation in the intensity, shape, and position of the CO stretching peak can be attributed to differences in hydrogen bonding, electrostatic interactions, and protonation effects when PANI/PAAMPSA interacts with the dopants. The control sample (Fig. S4) show a distinct peak at 1638 cm−1, which is assigned to the amide I CO vibrations of the PAAMPSA backbone49,50 shown in Fig. S5. A weak shoulder peak at 1723 cm−1 may be attributed to a minor fraction of free (non-hydrogen-bonded) carbonyl groups within the polymer matrix.51 The OH stretching band located at 3100–3600 cm−1 indicates hydrogen bonding between PANI, PAAMPSA, and the carboxylic-acid-based dopants.30
 |
| | Fig. 2 FTIR spectra of PANI/PAAMPSA/BA, PANI/PAAMPSA/CA, and PANI/PAAMPSA/DA. | |
2.3 Particle size and ζ potential
Particle size distribution (Fig. 3a) was analyzed because the size and uniformity of particles within a dispersion have a great influence on the processing and stability of the resulting polymer film.52 The PANI/PAAMPSA/BA film showed a smaller particle size of ∼1700 nm, with a narrow particle size distribution. However, both PANI/PAAMSA/CA and PANI/PAAMPSA/DA showed a notable shift toward higher particle sizes. PANI/PAAMSA/CA demonstrated a particle size of ∼3800 nm and PANI/PAAMSA/DA around ∼4100 nm. The larger particle size of PANI/PAAMSA/DA can be ascribed because the bulky, non-planar structure of the DA core increases the rigidity of the polymer network, creating steric hindrance. This prevents the dense packing, resulting in a larger hydrodynamic radius. In the case of CA, the higher particle size in comparison to that of PANI/PAAMSA/BA is likely due to the extensive hydrogen bonding from the tricarboxylate and hydroxyl functionalities. Particle size measurements clearly indicate that the dopant structure influences the interactions with PANI/PAAMPSA, which ultimately affects the colloidal stability. ζ-Potential determination was carried out at three different concentrations and is shown in Fig. 3b to determine the stability of the solution. All three dopants exhibited exceptionally high ζ-potential values above ∼–75 mV across the tested concentrations of 0.05, 0.3, and 1.5 w/v%, indicating strong colloidal stability.30 Comparable ζ potential values were observed for PANI/PAAMPSA/BA at all three concentrations. This consistent stability across these concentrations can be attributed to the narrow particle size distribution of PANI/PAAMPSA/BA. On the other hand, PANI/PAAMPSA/CA showed a linear increase in ζ potential with decreasing concentration. A very high ζ potential of ∼–130 mV is observed at 0.05 w/v% indicating high stability at this concentration. The decreased stability of PANI/PAAMPSA/DA at 1.5% is attributed to a combination of charge screening effects and interparticle association. At 1.5% the PANI/PAAMPSA/DA sample exhibits a ζ potential value of −75.9 mV, indicating the particles are negatively charged. However, at higher DA concentrations the ionized carboxylate groups increase the ionic strength and this leads to a compression of the electrical double layer surrounding the negatively charged particles. According to Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, when electrostatic repulsion is weakened, van der waals attractive forces may dominate, allowing the particles to approach more closely and potentially leading to aggregation.53 The presence of multiple acid functionalities and aromatic moieties may further promote intermolecular interactions, which can facilitate aggregation. Therefore the reduced stability at 1.5% DA likely arises from the combined influence of charge screening effects and interparticle association. Comparable ζ potential values of ∼–96, –97 mV were measured at both 0.3 w/v% and 0.05 w/v%, indicating highly stable dispersions.
 |
| | Fig. 3 (a) Particle size distribution of PANI/PAAMPSA/BA, PANI/PAAMPSA/CA, and PANI/PAAMPSA/DA. (b) ζ-Potential values of PANI/PAAMPSA/BA, PANI/PAAMPSA/CA, and PANI/PAAMPSA/DA at different concentrations 0.05%, 0.3% and 1.5%. The data plotted represents the mean and standard deviation (n = 3, n being the number of independent experiments). | |
2.4 UV-visible spectroscopy
The presence of PANI in its emeraldine salt form can be observed using the UV-vis absorption spectroscopy as shown in Fig. 4a. The schematic illustration of electronic transitions is given in the Fig. 4b. Three characteristic absorption peaks can be observed in the absorption spectrum: ∼350 nm, ∼420 nm and 760–800 nm. The absorption peak at ∼350 nm corresponds to the π–π* electron transition of benzenoid rings. This peak can be present in all PANI forms including emeraldine base, emeraldine salt, leucoemeraldine, and pernigraniline. The polaron-π* state transition was represented by the absorption peak at ∼420 nm. This peak represents the transition from polaron band to π* band.54 The three dopants showed similar peak values at the π–π* and polaron-π* transitions. However, the dopants showed different peak values at π-polaron transitions. The PANI/PAAMPSA/CA exhibited the absorption peak at 775 nm, PANI/PAAMPSA/BA at 790 nm, and PANI/PAAMPSA/DA at 765 nm. These peaks indicate the doped state of quinoid cations. The extent of the doping level can be estimated from the absorbance ratio of π-polaron to π–π* transition.55 The higher absorbance ratio level indicates more charge carriers, resulting in higher doping levels. Table 1 shows the UV-vis characteristic peak assignments and absorbance ratio of three dopants with PANI/PAAMPSA. The absorption ratio values indicate that both PANI/PAMPSA/CA and PANI/PAAMPSA/BA showed a higher degree of doping, followed by PANI/PAAMPSA/DA. The higher doping efficiency of PANI/PAAMPSA/BA might come from the multiple protonation sites and strong π–π stacking with PANI chains. In the case of PANI/PAAMPSA/CA, might be getting sufficient protonation through its three carboxylic acid groups. The bulky biphenyl structure of PANI/PAAMPSA/DA introduces steric hindrance. This disrupts PANI's planar backbone and limits both protonation efficiency and charge delocalization. Despite the stronger absorbances, the number of polarons formed during doping is significantly lesser for PANI/PAAMPSA/DA, resulting in a lower absorbance ratio.
 |
| | Fig. 4 (a) UV-vis absorption spectra of PANI/PAAMPSA/CABDs (b) schematic representation of the corresponding electronic transitions. | |
Table 1 UV-vis absorption peak assignments and absorbance ratios for PANI/PAAMPSA films with different CABDs
| Dopant |
π–π* Transition |
π-Polaron transition |
Absorbance ratio |
| (nm) |
Absorbance |
(nm) |
Absorbance |
| PANI/PAAMPSA/BA |
350 |
0.89 |
795 |
1.40 |
1.57 |
| PANI/PAAMPSA/CA |
350 |
0.70 |
775 |
1.10 |
1.57 |
| PANI/PAAMPSA/DA |
350 |
1.04 |
765 |
1.80 |
1.28 |
2.5 Conductivity measurements
The conductivity of PANI/PAAMPSA/CABDs depends on several factors such as dopant type, doping reaction, etc. If the structure is highly organized, electrons are easily conducted between the polymeric chains and the conductivity increases.45 Electrical conductivity (σ) of the films was calculated from the measured resistance values using eqn (1), where L is the distance between the electrodes (m), A is the cross-sectional area of the sample (m2), and R is the measured resistance (Ω).56 The dimensions of each sample were recorded prior to resistance measurements to ensure accurate determination of A.| | 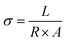 | (1) |
Conductivity values are given in Table 2. Higher conductivity is observed for PANI/PAAMPSA/BA films mostly due to high doping effciency, and the small size results in an orderly packing and π–π stacking with PANI's benzenoid/quinoid rings. PANI/PAAMPSA/CA showed conductivities slightly lower than those of PANI/PAAMPSA/BA. The fully aliphatic and highly hydrophilic nature contributes to the conductivity by providing a good protonation ability. The lowest conductivity of PANI/PAAMPSA/DA might be due to the lower protonation sites of DA compared to those of BA and CA. In addition, the bulky rigid hydrophobic structure reduces efficient π–π stacking with PANI, reducing conductivity. In conclusion, the larger particle size and aggregation of PANI/PAAMPSA/DA disrupt the percolation pathways, thereby reducing carrier mobility.
Table 2 Conductivity of PANI/PAAMPSA films with different dopants
| Dopants |
Conductivity (S m−1) |
| PANI/PAAMPSA/BA |
0.0063 |
| PANI/PAAMPSA/CA |
0.0059 |
| PANI/PAAMPSA/DA |
0.0021 |
2.6 Mechanical properties
The stress–strain graph shown in Fig. 5 compares the mechanical performance of PANI/PAAMPSA/BA,/PANI/PAAMPSA/CA, and PANI/PAAMPSA/DA. Table S3 gives the young's modulus, ultimate strength, maximum strength, and the toughness of the material. Young's modulus is the measure of stiffness in the material and is determined by the slope of the elastic region in the stress–strain curve.57 Toughness is the energy absorbed per unit volume before fracture, calculated from the area under the stress–strain curve. The maximum stress is the highest stress on the curve and the ultimate stress is the final peak stress just before fracture.CA-doped film demonstrated exceptional elongation of 3823% and the specimen did not fracture, but instead reached the maximum extension limit of the tensile testing machine. Consequently, because the film did not break, the true toughness of the film could not be determined. However, the toughness value calculated in the measurable strain range (6.11 MJ m−3) still provides a valid basis for comparison. Although this value does not represent the ultimate toughness due to instrument limitations, it confirms that the energy-absorbing capacity of the CA-doped film is substantially higher than that of the other dopants. The film exhibited a maximum tensile stress of 0.31 MPa and an ultimate stress of 0.30 MPa. This film exhibited a less young's modulus value (0.432 MPa) compared to the other dopants, concluding a lower stiffness due to the flexible chains. In the stress–strain graph, the dopant followed an elastic region from 0–150% of strain. After that, the material enters a plateau region where stress stays nearly constant or increases slightly. The hydrogen bond in the material breaks and reforms through molecular reorganization in this region. During the strain-hardening stage, the hydrogen bonds are stretched to their limits and network becomes stiffer.58 The multiple small peaks in PANI/PAAMPSA/CA can be attributed to the temporary stifness developed during the film's reformation, which caused localized increases in stress, followed by softening as the dynamic bonds subsequently broke and reformed.59 The lack of aromaticity and π–π stacking in CA-doped films imparts enhanced chain mobility, allowing the material to undergo breakage more easily while promoting efficient reformation.60The PANI/PAAMPSA/BA film exhibited the lowest elongation of 742% and the highest young's modulus of 3.08 MPa among the tested dopants. The aromatic ring of BA facilitates π–π stacking interactions, promoting a more locally ordered molecular arrangement. This enhanced stacking increases the energy required to disrupt intermolecular interactions and results in greater stiffness, which ultimately restricts chain mobility and limits the extensibility of the film. The PANI/PAAMPSA/DA film, on the other hand, exhibited moderate elongation, toughness, and young's modulus, while achieving a relatively high maximum stress. Despite having a highly rigid structure, the DA-doped film still demonstrated greater elongation than the BA-doped films. The mechanical properties of PANI/PAAMPSA films doped with different carboxylic acid-based dopants are summarized in Table 3.
 |
| | Fig. 5 Stress–strain curves of PANI/PAAMPSA/BA, PANI/PAAMPSA/CA, and PANI/PAAMPSA/DA. | |
Table 3 Mechanical properties of PANI/PAAMPSA films doped with different CABDs
| Sample |
Young's modulus (MPa) |
Maximum stress (MPa) |
Ultimate stress (MPa) |
Toughness (mJ m−3) |
| PANI/PAAMPSA/BA |
3.08 |
0.383 |
0.383 |
2.30 |
| PANI/PAAMPSA/CA |
0.432 |
0.315 |
0.300 |
6.11 |
| PANI/PAAMPSA/DA |
1.026 |
0.394 |
0.394 |
5.32 |
2.7 Thermal analysis
TGA and DSC analysis was performed to evaluate the thermal stability and thermal behavior of the prepared films. The TGA graph is shown in the Fig. 6a. Three stages of degradation were observed for all three dopants. During the initial stage between 30–190 °C the weight loss is caused by retained water, oligomers, free acids, residual initiator APS etc. The weight loss in the second stage from 190–320 °C is due to the loss of cross-linked dopants, main chain distortion, and decomposition of PAAMPSA side-chains. Furthermore, the molecular interactions (i.e., hydrogen bonding and dipole–dipole bonding) among the dopants, PAAMPSA, and PANI are dissipated in this step. The final weight loss step is from 320 to 780 °C, where the carbonization of the material occurs.61 The variation in the retained water is significant for the PANI/PAAMPSA system, since the water acts as a plasticizer. The retained water for each dopant is shown in the Fig. S6–S8. The PANI/PAAMPSA/CA sample exhibited the highest retained water of 15.1%, followed by PANI/PAAMPSA/DA (14.9%) and PANI/PAAMPSA/BA (10.5%). The higher retained water of PANI/PAAMPSA/CA is consistent with the hydrophilic nature of citric acid and the resulted high stretchability from the tensile studies. The tighter packing of PANI/PAAMPSA/BA creates a rigid network with a smaller free volume, resulting in the lowest water uptake and stretchability. PANI/PAAMPSA/DA possesses a more rigid core compared to BA. However, DA core prevents the efficient packing, leading to more disordered morphology with larger voids. This results in high retained water and greater deformation for PANI/PAAMPSA/DA film, compared to PANI/PAAMPSA/BA. In DSC thermogram Fig. 6b the peak at 120–125 °C can be attributed to the evaporation of retained water. The peak at 160–175 °C can be attributed to the small molecular-weight oligomers, and the peak at 175–180 °C can be assigned to the small molecule dopants. The endothermic peak at 200 °C can be ascribed to the thermal decomposition of the PAAMPSA side chains, while the peak at 312 °C is attributed to the onset of the polymer (PAAMPSA and PANI) main-chain decomposition and carbonization.30 The irregular peaks observed between 130–180 °C may be due to the loss of bound-water, which induces local expansion within the matrix and creates a more porous or hollow-like structure.
 |
| | Fig. 6 Thermal analysis of PANI/PAAMPSA films doped with carboxylic acid-based dopants (a) TGA curves recorded from 30–780 °C under N2 atmosphere (b) DSC thermograms recorded from 30–400 °C. | |
2.8 Mechanical and electrical conductivity self-healing studies
The self-healing efficiencies of the PANI/PAAMPSA/CABDs films were calculated based on electrical conductivity and mechanical stretchability, as defined in eqn (2) and (3). Here, S·H%c and S·H%s denote the self-healing efficiencies with regard to conductivity and stretchability, respectively. In these equations, σc, ε represent the initial conductivity and elongation at the break of the pristine film, while σc, ε correspond to the conductivity and elongation at break post-self-healing| | 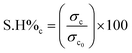 | (2) |
| | 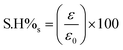 | (3) |
For the self-healing studies, each film was sliced in half using a sharp razor blade and the cut surfaces were carefully aligned to promote autonomous self-healing.62,63 The halves were gently brought into contact on separate glass slides to obtain maximum contact, without any misalignments or minor gaps, as these may negatively impact the healing process. The self-healing process is completely autonomous, as this process proceeds without the use of external stimuli or healing agents.
By the strain-based metric (S·H%s) the PANI/PAAMPSA/BA showed the highest healing efficiency at 46% (Fig. 7a), followed by PANI/PAAMPSA/CA (40%) (Fig. 7b) and PANI/PAAMPSA/DA (7.2%) (Fig. 7c). The S·H%c was highest for PANI/PAAMPSA/CA at 100% healing efficiency, followed by PANI/PAAMPSA/DA at 97% and PANI/PAAMPSA/BA at 72% (Fig. 7d). The difference in mechanical and electrical self-healing efficiencies highlights the different recovery mechanisms in PANI/PAAMPSA/CABDs films. The CA-doped film exhibited a full electrical recovery and a moderate mechanical recovery. Because of the hydrophilic structure of CA, the absorbed water provides a continuous proton–conduction medium that quickly re-establishes across the crack. Additionally, enhanced chain mobility in the film enables the reestablishment of polaron/bipolaron transport along the PANI backbone. In contrast, the weaker, less directional hydrogen bonds or ionic cross-links do not reform post-damage, limiting the mechanical recovery. Unlike BA/DA, CA lacks an aromatic motiff capable of π–π stacking, thereby reducing orderly contact points. PANI/PAAMPSA/BA film showed the highest mechanical recovery of 46% and a moderate electrical recovery of 72%. This behavior can be attributed to the orderly packing in the film, while its three –COOH anchor groups form strong, directional interactions that help in the reform process. The mechanical reconnecting mechanism of BA-doped film is more effective than the CA's or DA's. In contrast, the rigidity of these domains and their lower retained water limit proton-mediated transport, resulting in lower electrical recovery. In the case of DA, the rigid aromatic biphenyl structure carries high stress, limits effective π–π overlaps, relative to the more orderly packing of BA. As a result, the mechanical recovery is lower with moderate electrical recovery. Proton transport pathways within the network are more readily restored than bulk mechanical continuity, highlighting the importance of distinguishing between electrical and mechanical self-healing when evaluating conductive polymer systems.
 |
| | Fig. 7 Mechanical self-healing performance of (a) PANI/PAAMPSA/BA, (b) PANI/PAAMPSA/CA, and (c) PANI/PAAMPSA/DA showing representative stress–strain curves before healing and after self-healing. (d) Conductivity-based self-healing efficiencies of PANI/PAAMPSA/CABDs after 3 hours. The data plotted represents the mean and standard deviation (n = 3, n means number of independent experiments). | |
2.9 Re-processibility
Beyond their excellent mechanical properties and self-healability, these films are also fully re-processable. Fig. 8a–c shows the elongation-at-break curves collected before re-processing (B-RP) and after the first, second, and third re-processing cycles (RP-1, RP-2, RP-3). All three films were fully re-processable without loss of mechanical performance. BA-doped film showed an increase in elongation post-re-processing. Fig. 8d–f illustrates self-healing behavior after re-processing. Following re-processing, the self-healed PANI/PAAMPSA/BA films exhibit a decline in both strength and elongation. This can be due to a decrease in π–π domains, making the film more amorphous, allowing for greater elongation with lower mechanical recovery. The elongation and mechanical recovery of PANI/PAAMPSA/CA and PANI/PAAMPSA/DA remained nearly the same. From the re-processibility it is evident that the CABD films can be re-processed without loss of mechanical properties. In addition, the conductivities remained the same after re-processibility. The FTIR of the re-processed samples are shown in Fig. S9 and the graph shows no new bands or significant shift for PANI/PAAMPSA/BA (Fig. S9a), PANI/PAAMPSA/CA (Fig. S9b) and PANI/PAAMPSA/DA (Fig. S9c).
 |
| | Fig. 8 Mechanical performance of PANI/PAAMPSA composite films doped with different carboxylic acid-based dopants before and after reprocessibility and their subsequent self-healing ability. (a) Stress–strain curves of PANI/PAAMPSA/BA, (b) PANI/PAAMPSA/CA, and (c) PANI/PAAMPSA/DA films before reprocessing and after reprocessing (RP1–RP3). (d–f) Stress–strain curves of the same systems after reprocessing, showing self-healing behavior and recovery of mechanical properties. | |
2.10 Water degradability
The water degradability test was carried out using 1 cm × 1 cm specimen immersed in 10 ml of DI water, tap water and river water. To evaluate the sensor under realistic environmental conditions, the river water was sourced from the Penobscot river, which flows adjacent to the University of Maine campus Fig. 9a. Two experimental protocols were utilized to assess water degradability: (1) static immersion and (2) continuous stirring. Under continuous stirring, the three sensors were dissolved in each type of water in less than ten minutes. Notably, the dissolved solution was clear with no visible formation of sediments or insoluble contaminants, demonstrating the ability of the sensor to degrade in aqueous environment. However, under static immersion, a small portion of the sample remained intact, but upon gentle manual shaking, the samples dissolved, indicating that only a minimal energy is required to trigger rapid degradation. Also, the sensor can be disintegrated upon exposure to the slight physical forces, such as water flow present in natural water environments. Collectively, these results demonstrate environmentally relevant, water-transient behavior across different water sources. Fig. 9b illustrates the dissolution behavior of the three representative samples in Penobscot river water before and after stirring.
 |
| | Fig. 9 Water degradability of PANI/PAAMPSA/BA, PANI/PAAMPSA/CA, and PANI/PAAMPSA/DA films in river water under continuous stirring of 10 minutes (a) immediately after immersion and (b) after complete dispersion of the films. | |
2.11 Soil degradability
Soil degradability tests were conducted by placing the samples in the soil at room temperature.64 The pristine samples were solid prior to burial (Fig. 10a). After burial, the samples were softened, liquefied, and merged with the soil matrix, resulting in the complete loss of mechanical integrity after 24 hours. The Fig. 10b shows the samples integrated with the soil, indicating the degradability. The mass loss could not be calculated because the samples could not be taken from the soil. FTIR spectra before and after soil degradation are given in the Fig. S10a (PANI/PAAMPSA/BA), Fig. S10b (PANI/PAAMPSA/CA), Fig. S10c (PANI/PAAMPSA/DA). The overall FTIR spectral features before and after soil exposure remained largely similar. However, a noticeable reduction in peak intensities was observed after degradation. For all carboxylic acid doped films the SO and B–NH+ = Q bands merged into a broader peak after soil degradation. The quinoid band of PANI/PAAMPSA/BA film shifted from 1560 cm−1 to 1553 cm−1. Moreover, the carboxylic acid CO stretching band became undetectable after degradation, suggesting cleavage of carboxylic functionalities. Quantitative integration of the broad O–H stretching band (3800–2600 cm−1) showed a pronounced increase in intensity after soil degradation, with the integrated area increasing by approximately 124% for BA-doped films, 116% for CA-doped films, and 60% for DA-doped films. The increase in intensity is possibly due to the additional hydration from the soil. The SO3H peak area decreases by approximately 92.5% (BA), 92.1% (CA), and 83.7% (DA) after soil exposure, indicating substantial loss of sulfonic acid functionality across all systems, with BA and CA exhibiting the most pronounced degradation.
 |
| | Fig. 10 Soil degradation of PANI/PAAMPSA/CABDs (a) initial placement of PANI/PAAMPSA/BA, PANI/PAAMPSA/CA, and PANI/PAAMPSA/DA films in soil (b) films after 24 h of soil burial showing visible degradation and fragmentation. | |
3 Conclusion
In conclusion, three carboxylic acid based dopants: 1,2,4-benzene tricarboxylic acid, citric acid, and diphenic acid and their effects on doping with PANI/PAAMPSA are evaluated in this study. These dopants are structurally varied by their nature and number of acidic groups, presence of benzene or biphenyl rings, and inherent structural flexibility. The flexible, hydrophilic structure of CA-doped PANI/PAAMPSA enabled a high elongation of 3823%, retained water of 15.1% and exhibited 100% recovery of electrical conductivity upon self-healing. In contrast, the more orderly packing of BA-doped PANI/PAAMPSA films resulted in the highest conductivity, increased Young's modulus, and enhanced mechanical self-healing efficiency. A comparable doping efficiency of 1.57 was achieved for both PANI/PAAMPSA/BA and PANI/PAAMPSA/CA films. PANI/PAAMPSA/DA film, with its highly rigid biphenyl structure, demonstrated only moderate recovery in both electrical conductivity and mechanical self-healing. All three films exhibited high re-processibility, allowing them to be dissolved in water and reformed into new films upon drying. Water degradation studies demonstrated rapid dissolution of the films in tap water, deionized water, and river water within 10 minutes, underscoring their transient nature. In addition, soil burial tests showed complete degradation within 24 hours, further confirming their environmental sustainability. Our results reveal a clear trade-off, in which BA-doped films offer the highest electrical conductivity, CA-doped films excel in stretchability and electrical self-healing, and DA-doped films exhibit moderate electrical and mechanical performance, while all systems maintain comparable degradability. Hence, the dopants can be selected based on application-specific requirements, balancing conductivity, stretchability, and degradability to meet targeted device needs. Together, these findings demonstrate that carboxylic acid-doped PANI/PAAMPSA films shows high performance with rapid environmental degradability and re-processibility, making them promising candidates for sustainable, transient electronics applications, particularly in soft robotics, artificial skin, and flexible bioelectronics.
Author contributions
Conceptualization, funding acquisition, and project administration was carried out by E. K. W. Resources were provided by C. D. Investigation, visualization, and original draft writing was done by A. A and T. W. Methodology was conducted by C. D., T. W., and A. A. Reviewing & editing of the manuscript was carried out by A. A., C. D., T. W., and E. K. W.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Further data will be made available upon request. Supplementary information is available. See DOI: https://doi.org/10.1039/d5ta10370c.
Acknowledgements
E. Wujcik and A. Ajeev would like to acknowledge support from a National Science Foundation (NSF) CAREER Award from the Electronic/Photonic Materials program (NSF/MPS/DMR/EPM; award: 2305282).
Notes and references
- D. Davis, S. K. Narayanan, A. Ajeev, J. Nair, J. Jeeji, A. Vijayan, M. Viyyur Kuttyadi, A. Nelliparambil Sathian and A. K. Arulraj, ACS Appl. Mater. Interfaces, 2023, 15, 25734–25743 CrossRef CAS PubMed.
- J. Xiong, J. Chen and P. S. Lee, Adv. Mater., 2021, 33, 2002640 CrossRef CAS PubMed.
- C. Duprey, A. Ajeev, D. Hong, K. Webb, S. Veres, G. Chen, E. Linn, G. Lusvardi, Z. Liu and R. Wang,
et al.
, Adv. Compos. Hybrid Mater., 2025, 8, 1–19 Search PubMed.
- H. Dong, X. Li, Y. Liu, W. Cheng, X. Li, D. Lu, C. Shao and Y. Liu, ACS Sens., 2024, 9, 3085–3095 CrossRef CAS PubMed.
- C. Duprey, H. Rouhi, S. Alipoori, L. McLoughlin, S. Veres, G. Chen, C. Cook, O. Diaz, E. Linn and Y. Lu,
et al.
, ES energy environ., 2023, 22, 962 CAS.
- H. Rouhi, C. Duprey, C. Cook, E. Linn, S. Veres, G. Chen, A. Alshaikh, Y. Lu, L. Terry and M. Elliott,
et al.
, J. Appl. Polym. Sci., 2023, 140, e53294 CrossRef CAS.
- Y. Lu, G. Yu, X. Wei, C. Zhan, J.-W. Jeon, X. Wang, C. Jeffryes, Z. Guo, S. Wei and E. K. Wujcik, Adv. Compos. Hybrid Mater., 2019, 2, 711–719 CrossRef CAS.
- A. Ajeev, B. H. Javaregowda, A. Ali, M. Modak, S. Patil, S. Khatua, M. Ramadoss, P. A. Kothavade and A. K. Arulraj, Adv. Mater. Technol., 2020, 5, 2000690 CrossRef.
- S. Li, Y. Wang, J. Xia, C. Zhuge, W. Lu and N. Li, ACS Appl. Polym. Mater., 2024, 6, 8756–8766 CrossRef CAS.
- A. Ajeev, T. Warfle, S. Maslaczynska-Salome, S. Alipoori, C. Duprey and E. K. Wujcik, Chem. Sci., 2025, 16, 9056–9075 RSC.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Chem. Mater., 2016, 28, 3527–3539 CrossRef CAS.
- A. H. Majeed, L. A. Mohammed, O. G. Hammoodi, S. Sehgal, M. A. Alheety, K. K. Saxena, S. A. Dadoosh, I. K. Mohammed, M. M. Jasim and N. U. Salmaan, Int. J. Polym. Sci., 2022, 2022, 9047554 Search PubMed.
- Z. A. Boeva and V. G. Sergeyev, Polym. Sci. Ser. C, 2014, 56, 144–153 CrossRef CAS.
- J. E. Yoo, T. L. Bucholz, S. Jung and Y.-L. Loo, J. Mater. Chem., 2008, 18, 3129–3135 RSC.
- P. B. D. Firda, Y. T. Malik, J. K. Oh, E. K. Wujcik and J.-W. Jeon, Polymers, 2021, 13, 2992 CrossRef CAS PubMed.
- Y. Ma, M. Ma, X. Yin, Q. Shao, N. Lu, Y. Feng, Y. Lu, E. K. Wujcik, X. Mai and C. Wang,
et al.
, Polymer, 2018, 156, 128–135 CrossRef CAS.
- A. Ajeev, C. Duprey and E. K. Wujcik, Chem. Commun., 2026, 62, 3560–3563 RSC.
- N. Sharma, A. Singh, N. Kumar, A. Tiwari, M. Lal and S. Arya, J. Mater. Sci., 2024, 59, 6206–6244 CrossRef CAS.
- H. Hussin, S. N. Gan, S. Mohamad and S. W. Phang, Polym. Polym. Compos., 2017, 25, 515–520 CAS.
- S. Maslaczynska-Salome, A. Ajeev, T. Warfle, B. Shams, C. Duprey and E. K. Wujcik, Macromol. Chem. Phys., 2026, 227, e00293 CrossRef CAS.
- M. Beygisangchin, A. H. Baghdadi, S. K. Kamarudin, S. A. Rashid, J. Jakmunee and N. Shaari, Eur. Polym. J., 2024, 210, 112948 CrossRef CAS.
- H. Wan, C. Qin and A. Lu, J. Mater. Chem. A, 2022, 10, 17279–17287 RSC.
- P. Rahmani, A. Shojaei and M. D. Dickey, J. Mater. Chem. A, 2024, 12, 9552–9562 RSC.
- X. Liu, Y. Yang, S. Song, R. Zhang, C. Zhang, S. Yang, Y. Liu and Y. Song, Int. J. Biol. Macromol., 2024, 280, 135959 CrossRef CAS PubMed.
- J. Wu, J. Xu, S. Liu, Z. Ma, Y. Guo and X. Wang, Int. J. Biol. Macromol., 2025, 306, 141662 CrossRef CAS PubMed.
- C. Liu, Y. Sun, P. Liu, F. Ma, S. Wu, J. Li, S. Li, R. Hu, Z. Wang and Y. Wang,
et al.
, Int. J. Biol. Macromol., 2023, 231, 123568 CrossRef CAS PubMed.
- H. Zhang, H. Shen, J. Lan, H. Wu, L. Wang and J. Zhou, Carbohydr. Polym., 2022, 295, 119848 CrossRef CAS PubMed.
- X. Zou, H. Liu, Z. Hu, Y. Zhang, J. Cheng, K. Wang, Y. Feng and J. Wang, Carbohydr. Polym., 2025, 354, 123342 CrossRef CAS PubMed.
- J. E. Yoo and J. Bae, Macromol. Res., 2015, 23, 749–754 CrossRef CAS.
- Y. Lu, Z. Liu, H. Yan, Q. Peng, R. Wang, M. E. Barkey, J.-W. Jeon and E. K. Wujcik, ACS Appl. Mater. Interfaces, 2019, 11, 20453–20464 CrossRef CAS PubMed.
- A. Ajeev, C. Duprey and E. K. Wujcik, ACS Appl. Polym. Mater., 2025, 7(21), 14562–14572 CrossRef CAS.
- M. Sezen-Edmonds, P. P. Khlyabich and Y.-L. Loo, ACS Appl. Mater. Interfaces, 2017, 9, 12766–12772 CrossRef CAS PubMed.
- M. Sezen-Edmonds and Y.-L. Loo, J. Phys. Chem. Lett., 2017, 8, 4530–4539 CrossRef CAS PubMed.
-
A. Sáenz-Galindo, L. I. López-López, N. Fabiola, A. O. Castañeda-Facio, L. A. Rámirez-Mendoza, K. C. Córdova-Cisneros and D. de Loera-Carrera, in Carboxylic acid: key role in life sciences, 2018, vol. 35, pp. 1–35 Search PubMed.
- E. M. Maya, J. Benavente and J. de Abajo, Mater. Chem. Phys., 2012, 131, 581–588 CrossRef CAS.
-
A. Göpferich, in The Biomaterials: Silver Jubilee Compendium, 1996, pp. 117–128 Search PubMed.
-
G. Odian, in Principles of Polymerization, John Wiley & Sons, Hoboken, NJ, 4th edn, 2004 Search PubMed.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
-
S. H. Hamid, in Handbook of Polymer Degradation, CRC Press, Boca Raton, FL, 2nd edn, 2000 Search PubMed.
- V. M. Pathak,
et al.
, Bioresour. bioprocess., 2017, 4, 1–31 Search PubMed.
- V. Suresh, R. Shams, K. K. Dash, A. M. Shaikh and K. Béla, J. Agric. Food Res., 2025, 101788 CAS.
- J. Choi, H. Kim, Y.-R. Ahn, M. Kim, S. Yu, N. Kim, S. Y. Lim, J.-A. Park, S.-J. Ha and K. S. Lim,
et al.
, RSC Adv., 2024, 14, 9943–9966 RSC.
- U. Rana, K. Chakrabarti and S. Malik, J. Mater. Chem., 2012, 22, 15665–15671 RSC.
- M. Charton, J. Org. Chem., 1977, 42, 2528–2529 CrossRef CAS.
- N. Nepomuceno, A. Seixas, E. Medeiros and T. Mélo, J. Solid State Chem., 2021, 302, 122372 CrossRef CAS.
- K. Bednarczyk, W. Matysiak, T. Tański, H. Janeczek, E. Schab-Balcerzak and M. Libera, Sci. Rep., 2021, 11, 7487 CrossRef CAS PubMed.
- J.-W. Jeon, J. O'Neal, L. Shao and J. L. Lutkenhaus, ACS Appl. Mater. Interfaces, 2013, 5, 10127–10136 CrossRef CAS PubMed.
- J. Stejskal, D. Hlavatá, P. Holler, M. Trchová, J. Prokeš and I. Sapurina, Polym. Int., 2004, 53, 294–300 CrossRef CAS.
- B. de Campos Vidal and M. L. S. Mello, Micron, 2011, 42, 283–289 CrossRef PubMed.
- E. Rogha, M. Bazli, M. Shakiba, A. Rajabipour, R. Hassanli, C. O. Ojo, G. Aryal and H. A. Campbell, Compos. Commun., 2025, 102591 CrossRef.
- K.-Y. Chiang, F. Matsumura, C.-C. Yu, D. Qi, Y. Nagata, M. Bonn and K. Meister, J. Phys. Chem. Lett., 2023, 14, 4949–4954 CrossRef CAS PubMed.
- J. Huang, H. Zhou, L. Zhang, L. Zhang, W. Shi, Y. Yang, J. Zhou, T. Zhao and M. Liu, Nat. Commun., 2024, 15, 6747 CrossRef CAS PubMed.
- Y. Liang, N. Hilal, P. Langston and V. Starov, Adv. Colloid Interface Sci., 2007, 134, 151–166 CrossRef PubMed.
- D. Y. Imali, E. C. J. Perera, M. Kaumal and D. P. Dissanayake, RSC Adv., 2023, 13, 6396–6411 RSC.
- H. Xia and Q. Wang, J. Nanopart. Res., 2001, 3, 399–409 CrossRef.
-
M. B. Heaney, in Electrical measurement, signal processing, and displays, 2003, vol. 7, pp. 1–14 Search PubMed.
- Z.-Q. Gao, C.-H. Liu, S.-L. Zhang, S.-H. Li, L.-W. Gao, R.-L. Chai, T.-Y. Zhou, X.-J. Ma, X. Li and S. Li,
et al.
, Small, 2024, 20, 2404231 CrossRef CAS PubMed.
- C. K. Locke and Y. Yang, Macromolecules, 2025, 58(12), 6326–6340 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- Y. Ren and X. Dong, Prog. Polym. Sci., 2024, 158, 101890 CrossRef CAS.
- M. F. Aizamddin, Z. Zainal Ariffin, N. A. Nor Amdan, M. A. Nawawi, N. A. Jani, M. F. Safian, S. N. A. Shaffee, N. M. R. Nik Mohamed Daud, M. M. Myo Thant and M. M. Mahat, ACS Omega, 2024, 9, 23303–23315 CrossRef CAS PubMed.
- C.-Z. Hang, X.-F. Zhao, S.-Y. Xi, Y.-H. Shang, K.-P. Yuan, F. Yang, Q.-G. Wang, J.-C. Wang, D. W. Zhang and H.-L. Lu, Nano Energy, 2020, 76, 105064 CrossRef CAS.
- Z. Zhang, Z. Gao, Y. Wang, L. Guo, C. Yin, X. Zhang, J. Hao, G. Zhang and L. Chen, Macromolecules, 2019, 52, 2531–2541 CrossRef CAS.
- B. Shams, D. W. Bousfield and E. K. Wujcik, Composites, Part B, 2025, 112549 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab












