DOI:
10.1039/D5TA10237E
(Paper)
J. Mater. Chem. A, 2026,
14, 14214-14223
Synergistic tailoring of ion–dipole interactions and segmental dynamics in fluorinated ionogels for low-temperature micro-supercapacitors
Received
16th December 2025
, Accepted 26th February 2026
First published on 27th February 2026
Abstract
Polymeric gel electrolytes offer stable ion-transport under mechanical deformation, enabling reliable operation in wearable and flexible energy-storage devices. However, their performance deteriorates notably at low-temperatures due to reduced ionicity and restricted molecular motion within the polymer network. Addressing this challenge requires a rational gel electrolyte design that promotes ion dissociation and transport even at low-temperatures. Here, we propose a synergistic molecular tailoring strategy for copolymer gelators. Fluorinated functional groups are introduced to attenuate the local electrostatic environment, facilitating the dissociation of ion pairs, while side-chain extension creates a more compliant and less temperature-dependent network that preserves ion motion at low-temperatures. Incorporating our ionogel into micro-supercapacitors enables stable electrochemical operation at temperatures more than 30 °C below the limit of typical systems, while maintaining cycling stability. Overall, these results demonstrate that molecularly tailored copolymer networks can effectively mitigate the intrinsic temperature sensitivity of gel electrolytes and establish a broadly applicable design principle for energy-storage devices operating in extreme environments.
Introduction
Reliable energy storage in extreme environmental conditions, such as polar regions or outer space, requires electrolytes that can maintain high ionic conductivity at low temperatures.1–3 Concurrently, the increasing deployment of wearable, portable, and structurally adaptive devices demands energy-storage systems that can tolerate bending, stretching, and other forms of mechanical deformation.4–6 Satisfying both requirements is difficult with liquid electrolytes. Their fluidity causes leakage risks, and their ionic conductivity deteriorates severely in cold conditions.7 Polymeric gel electrolytes offer a promising alternative, as their mechanically compliant networks provide continuous ion-transport pathways suitable for deformable energy-storage systems.8,9 However, gels containing ionic liquids or organic solvents suffer from freezing-related limitations, which obstruct ion-transport and degrade device performance.10–13
Previous studies on low-temperature electrolytes have primarily utilized antifreezing additives to overcome this challenge. Co-solvents are widely employed as antifreezing additives, as they modify the hydrogen-bonded water network and lower the chemical potential of water. For example, water freezing in hydrogels is effectively prevented by adding co-solvents such as dimethyl sulfoxide,14 glycerol,15 and ethylene glycol.16 Another representative approach involves the use of natural biomolecules (e.g., ice-binding proteins, antifreeze proteins, and glycoproteins) that inhibit ice recrystallization.17–19 By adsorbing onto ice surfaces, these additives perturb the local organization of solvent molecules to suppress crystal growth and recrystallization. Despite their effectiveness, additive-based strategies often induce phase separation20,21 and increased viscosity22,23 in gel electrolytes, limiting their use in energy storage devices. Therefore, developing a fundamentally new route that overcomes the limitations of conventional approaches and intrinsically regulates ionic behavior in gel electrolytes remains a critical challenge.
In this study, we propose a synergistic molecular design strategy for copolymer gelators that not only enhances ionic conductivity but also reduces their temperature sensitivity. The incorporation of fluorinated side groups progressively strengthens ion–dipole interactions, increasing the loose ion-pair fraction from about 36% to 63%. Furthermore, a more compliant network lowers the rheological activation energy from 90.3 to 20.5 kJ mol−1, implying a significant decrease in the temperature sensitivity. When the optimized gel was applied to micro-supercapacitors (MSCs), an areal capacitance of 2.04 mF cm−2 was achieved at 25 °C. These MSCs retained 97% of their capacitance after 1000 bending cycles, demonstrating durability under repeated deformation. Also, the device exhibited reliable cycling stability with 95% retention of its initial capacitance after 5000 charge–discharge cycles. Moreover, MSCs incorporating fluorinated copolymer gelators remained successfully operable at temperatures more than 30 °C below their typical operating range and maintained stable performance under low-temperature conditions. Overall, this molecular design offers a generalized strategy for mitigating the temperature sensitivity of polymeric gel electrolytes and enabling stable energy-storage performance in extreme environments.
Results and discussion
Synergistic tailoring of copolymer gelators
Copolymer gelators used in ionogels consist of ionic liquid phobic (IL-phobic) and ionic liquid philic (IL-philic) domains.24–26 The IL-phobic domains provide mechanical robustness, while the IL-philic domains govern the ion-transport characteristics. We fixed polystyrene (PS) as the IL-phobic domain and varied only the IL-philic domain. Since ion-transport is closely tied to the IL-philic domain, our modifications were focused on this domain. Three copolymer gelators—poly(styrene-ran-ethyl methacrylate) (PS-r-PEMA), poly(styrene-ran-trifluoroethyl methacrylate) (PS-r-PtFEMA), and poly(styrene-ran-octafluoropentyl methacrylate) (PS-r-PoFPMA)—were synthesized by tailoring the IL-philic domains through variations in fluorination and side-chain length (Fig. 1a and S1). Proton nuclear magnetic resonance (1H NMR) spectroscopy and gel permeation chromatography (GPC) confirmed the synthesis of the copolymer gelators with consistent styrene ratios and comparable molecular weights (Fig. S2 and S3). The synthesized copolymer gelators were combined with 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([BMI][TFSI]) to form ionogels, denoted as PEMA ionogel (from PS-r-PEMA), PtFEMA ionogel (from PS-r-PtFEMA), and PoFPMA ionogel (from PS-r-PoFPMA), respectively. The composition of the ionogels was optimized by evaluating both the viscoelastic properties and ionic conductivities of PoFPMA ionogels with varying polymer to ionic liquid ratios (Fig. S4). The 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio demonstrated superior ionic conductivity while preserving the gel morphology. Using the optimized composition, we prepared PEMA, PtFEMA, and PoFPMA ionogels, each showing a gel-like viscoelastic response (Fig. S5).
:6 ratio demonstrated superior ionic conductivity while preserving the gel morphology. Using the optimized composition, we prepared PEMA, PtFEMA, and PoFPMA ionogels, each showing a gel-like viscoelastic response (Fig. S5).
This synergistic strategy consists of two complementary approaches. First, the ion carrier enhancement strategy increases the number of mobile ion carriers (n). Second, the optimization of ionogel dynamics enhances ion mobility (µ). Consequently, these concurrent improvements lead to enhanced ionic conductivity (σ), as described by the Nernst–Einstein equation (σ = nqµ) (Fig. 1b).27 The schematic illustration in Fig. 1c conceptually summarizes how the tailored IL-philic domains promote ion dissociation and increase chain mobility in ionogels. In the PEMA ionogel, non-fluorinated side-chains engage in weak ion–dipole interactions with the cation of the ionic liquid, which results in limited ion dissociation.28 In the PtFEMA ionogel, the partially fluorinated side-chains moderately strengthen the ion–dipole interactions, which increases the number of dissociated ion carriers.29,30 In addition, the partial fluorination creates a more favorable enthalpic interaction with the ionic liquid, improving its miscibility within the polymer matrix.31 This enhanced miscibility leads to increased chain mobility. In the PoFPMA ionogel, the highly fluorinated side-chains most strongly promote ion dissociation by further strengthening the ion–dipole interactions. Additionally, the extensive fluorination further stabilizes the ionic liquid within the polymer matrix, and the elongated side-chains contribute to higher chain mobility. Overall, the combination of extensive fluorination and extended side-chains most effectively enhances both ion dissociation and chain mobility, yielding the highest ion dynamics in the PoFPMA ionogel.
 |
| | Fig. 1 Synergistic polymer engineering strategy for enhancing ionicity and ion mobility in ionogels. (a) Modulation of copolymer gelators: non-fluorinated short side-chain (PEMA ionogel), slightly fluorinated short side-chain (PtFEMA ionogel), and highly fluorinated long side-chain (PoFPMA ionogel). (b) Correlation between ionic conductivity (σ), ionic carrier concentration (n), and ion mobility (µ), demonstrating the conductivity improvement due to increased ion carrier and ion mobility. (c) Schematic illustration of ion dynamics within each ionogel system. | |
Effect of copolymer gelators on ion carrier enhancement
In gel electrolytes, greater ion dissociation results in a higher carrier concentration (n), leading to increased ionic conductivity.32 As the first strategy, the effect of side-chain fluorination on mobile ion carriers was evaluated by comparing PEMA, PtFEMA, and PoFPMA ionogels with progressively increasing fluorination. Fig. 2a presents the overall mechanism by which fluorinated side-chains promote ion dissociation: (1) the fluorinated moieties interact with the cation through enhanced ion–dipole interactions, (2) these interactions reduce the coulombic attraction to the anion, and (3) the weakened cation–anion coupling consequently increases the proportion of loose ion pairs. Ionic interactions in the ionogels were analyzed by Fourier-transform infrared (FTIR) spectroscopy and Raman spectroscopy. As shown in Fig. 2b, the C–H symmetric stretching peak of [BMI]+ gradually shifted to a lower wavenumber with increasing fluorine content, indicating enhanced interactions between the fluorinated moieties of the copolymer gelators and the cation. Similarly, the SNS and SO2 stretching peaks of [TFSI]− shifted to higher wavenumbers with increasing fluorination, which reflects a weakened cation–anion coulombic force in the ionic liquid (Fig. 2c and d).32,33 Furthermore, a progressive peak shift was observed with increasing polymer content, which confirms the strengthening of these effects (Fig. S6). These FTIR results reveal that higher fluorination on the side-chains improves ion–dipole interactions, which diminishes the coulombic force between the cation–anion pairs.
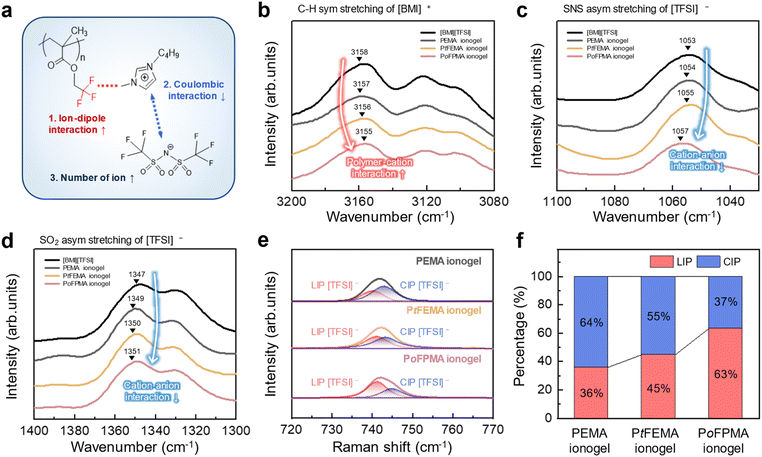 |
| | Fig. 2 Fluorine mediated increased ion carriers. (a) Schematic illustration of the fluorinated side-chain effects on the ionogels. (b–d) FTIR spectra illustrating (b) C–H symmetric stretching of [BMI]+, (c) SNS asymmetric stretching of [TFSI]−, and (d) SO2 asymmetric stretching of [TFSI]− for [BMI][TFSI] and the ionogels. (e) Deconvoluted Raman spectra of [TFSI]− for ionogels. (f) Relative fractions of LIP and CIP obtained from the Raman analysis. | |
Further analysis of the [TFSI]− coordination state was conducted using Raman spectroscopy on ionogels with different degrees of fluorination. In the Raman spectra, the [TFSI]− anion can be distinguished in two coordination states: a loose ion-paired (LIP) state, in which the [TFSI]− anion interacts weakly with the cation, and a contact ion-paired (CIP) state, where strong coulombic interactions occur between the cation and the [TFSI]− anion.34–36 In Fig. 2e, the Raman spectra were deconvoluted into LIP and CIP components, confirming the coexistence of both ion-pairing states in all ionogels. Quantitative analysis of the Raman spectra revealed a gradual increase in the fraction of LIP components from 36% in PEMA to 45% in PtFEMA and 63% in PoFPMA (Fig. 2f). Consequently, the fluorinated side-chains increase the number of mobile ion carriers by promoting the dissociation of ion pairs in the ionic liquid.
Ionogel dynamics and temperature sensitivity
As a second strategy, polymer chain mobility was tuned to modulate the viscoelastic behavior of the ionogels, creating a more dynamic network that promotes ion motion and enhances ion mobility (µ). Differential scanning calorimetry (DSC) was used to determine the glass transition temperatures (Tg) of the ionogels and their corresponding neat polymers (Fig. S7). Neat PEMA and PtFEMA show similar Tg values. However, a large difference in Tg is observed between their ionogels, indicating improved miscibility with the ionic liquid. Moreover, the further decrease in PoFPMA ionogels reflects enhanced miscibility and increased side-chain length. The relaxation dynamics of the PEMA, PtFEMA, and PoFPMA ionogels were examined through time–temperature superposition (tTS) analysis, using 20 °C as the reference temperature (Fig. 3a–c). The terminal relaxation point is defined by the crossover of G′ and G″, and the terminal relaxation time (τt) is determined from the reciprocal of the corresponding crossover frequency.37 This crossover point represents the transition between the terminal and plateau zones, reflecting the structural relaxation of the polymer network. The τt decreased markedly from 4.12 h for PEMA to 746 s for PtFEMA and 13 s for PoFPMA, indicating progressively boosted polymer network relaxation (Fig. S8). This accelerated network relaxation promotes faster electrochemical ion transport within the ionogels. The stress relaxation modulus (G(t)) decayed faster in PoFPMA than in PEMA and PtFEMA, reflecting a more rapid loss of stress over time (Fig. S9). Consequently, enhanced chain dynamics promote ion mobility within the ionogel network.
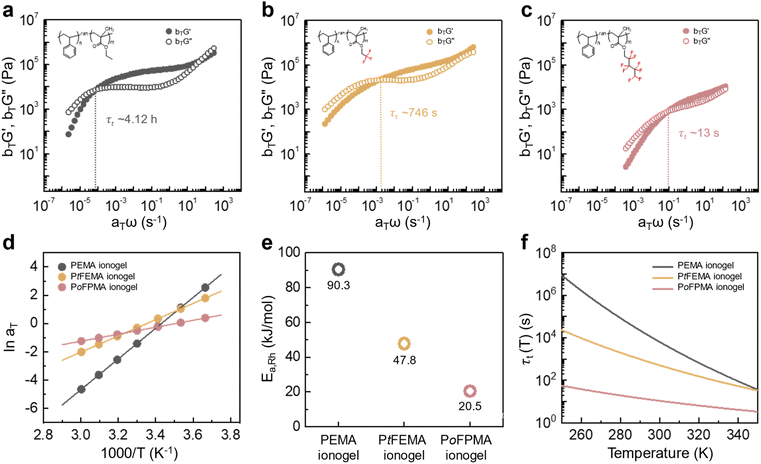 |
| | Fig. 3 Ionogel dynamics and temperature sensitivity. (a–c) Time–temperature superposition master curves of the (a) PEMA, (b) PtFEMA, and (c) PoFPMA ionogels. (d) Arrhenius plots of the temperature-dependent shift factors ln(aT) for the ionogels. (e) Comparison of activation energies (Ea,Rh) derived from the Arrhenius plots. (f) Calculated terminal relaxation time τt (T) of the PEMA, PtFEMA, and PoFPMA ionogels based on the Arrhenius fits. | |
The horizontal shifting factors (aT) were subsequently obtained from the tTS master curves to analyze the temperature dependence of the relaxation behavior (Table S1). These aT values were fitted with the Arrhenius equation (eqn (1)), derived from eqn (2), (3) and (Fig. 3d). From this analysis, the rheological activation energy (Ea,Rh) decreased from 90.3 kJ mol−1 for PEMA to 47.8 kJ mol−1 for PtFEMA and 20.5 kJ mol−1 for PoFPMA (Fig. 3e). This decrease indicates reduced temperature sensitivity of the ionogel dynamics. Based on the extracted activation energy and the experimentally measured τt, the temperature dependence of τt(T) for each ionogel was evaluated (Fig. 3f). The slope of the plot corresponds to the temperature sensitivity of τt, and PoFPMA exhibited the lowest value among the ionogels. This result implies that the ionogels with lower temperature sensitivity maintain stable viscoelastic relaxation regardless of temperature, enabling reliable performance under low-temperature conditions.
| | 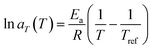 | (1) |
| | 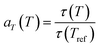 | (2) |
| | 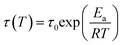 | (3) |
Electrochemical properties of ionogels
The effect of this synergistic design-enhancing both the number of charge carriers (n) and their mobility (µ)-on ionic conduction was examined using electrochemical impedance spectroscopy (EIS). This enhanced ionic conduction is anticipated to improve the electrochemical performance of the ionogels, particularly their ionic conductivity, capacitance, and relaxation behavior. The ionic conductivities of PEMA, PtFEMA, and PoFPMA are 0.110, 0.333, and 0.649 mS cm−1, demonstrating the progressive enhancement in ion dissociation and chain mobility (Fig. 4a). Bode plots measured at different temperatures (0–25 °C) were obtained for the three ionogels (Fig. 4b and S10). Activation energies for ionic conduction (Ea,EC) were determined from the Arrhenius plots (Fig. S11), yielding values of 52.4, 47.9, and 41.2 kJ mol−1 for PEMA, PtFEMA, and PoFPMA, respectively (Fig. 4c). The lowest activation energy in PoFPMA reflects its reduced ion-transport barrier. Ion behavior was further examined using Bode plots (Fig. 4d). The dielectric relaxation time (τD) was obtained from the reciprocal of the frequency at which the real (Z′) and imaginary (Z″) components intersect.38τD decreased from 61.7 to 14.2 and 6.6 ms for PEMA, PtFEMA, and PoFPMA, respectively, reflecting progressively faster charge relaxation (Fig. 4e). This result reveals the coupled relationship between mechanical relaxation and ion dynamics in the ionogels (Fig. S12). This coupled relationship indicates that the ion transport for EDL formation is governed by the polymer dynamics. Ion-transport differences were further analyzed through the frequency-dependent areal capacitance of the ionogels (Fig. 4f). The specific capacitance at 0.1 Hz increased from 12.9 to 15.2 and 18.2 µF cm−2 for PEMA, PtFEMA, and PoFPMA, respectively, demonstrating that enhanced ion-transport enables more efficient charge accumulation.39 Effective ion accumulation at the electrode interface leads to the formation of an EDL, which contributes to the enhanced capacitive performance of the capacitor. These results collectively indicate that the ionogels exhibit fast charge relaxation and capacitive behavior, demonstrating their potential applicability in energy-storage devices.
 |
| | Fig. 4 Electrochemical characterization of the ionogels. (a) Ionic conductivity (σ) of ionogels. (b) Bode plots of the PoFPMA ionogels measured at various temperatures. (c) Comparison of activation energies (Ea,EC) derived from the Arrhenius plots. (d) Real and imaginary impedance of the ionogels as a function of frequencies. (e) Dielectric relaxation time (τD) of ionogels. (f) Areal capacitance of the ionogels. | |
Performance of ionogel-based MSCs
The improved electrochemical properties of the ionogels were subsequently examined in MSC devices to evaluate their device-level applicability. The MSCs were fabricated by patterning laser-induced graphene (LIG) electrodes on a polyimide (PI) film using CO2 laser irradiation (Fig. 5a).40,41 The optical image of the fabricated LIG electrodes is shown in Fig. S13. The ionogels were subsequently drop-cast onto these electrodes. The formation of the LIG electrodes was confirmed through scanning electron microscopy (SEM) imaging and Raman spectroscopy (Fig. S14 and S15). Cyclic voltammetry (CV) was performed under various scan rates within a 1.5 V potential window to evaluate the charge storage behavior of the MSCs (Fig. 5b and S16). The CV curves maintained a nearly rectangular shape even at a high scan rate of 100 mV s−1, indicating efficient EDL formation. Fig. 5c and S17 show galvanostatic charge–discharge (GCD) curves measured at various current densities. All GCD curves exhibited symmetric triangular profiles, and the PoFPMA MSC showed the longest charging and discharging durations among the three ionogels. The areal capacitances of the MSCs were obtained from the GCD profiles at various current densities (Fig. 5d and S18). The PoFPMA MSC recorded the highest areal capacitance at 0.01 mA cm−2, delivering 2.04 mF cm−2, whereas the PtFEMA and PEMA MSCs showed 1.24 and 1.05 mF cm−2, respectively. The PoFPMA MSC exhibited the highest energy density of 0.425 µWh cm−2 at a power density of 5.01 mW cm−2 in the Ragone plots (Fig. 5e and S19). The GCD profiles remained nearly identical when the MSC was bent to radii of 2 cm and 1.5 cm, demonstrating the mechanical flexibility (Fig. 5f). The PoFPMA MSC retained 97% of its capacitance after 1000 bending cycles, indicating fatigue resistance under repeated deformation (Fig. 5g). The capacitance retention was maintained at 95% after 5000 charge–discharge cycles, confirming the long-term stability of the PoFPMA MSC (Fig. 5h and Table S2).
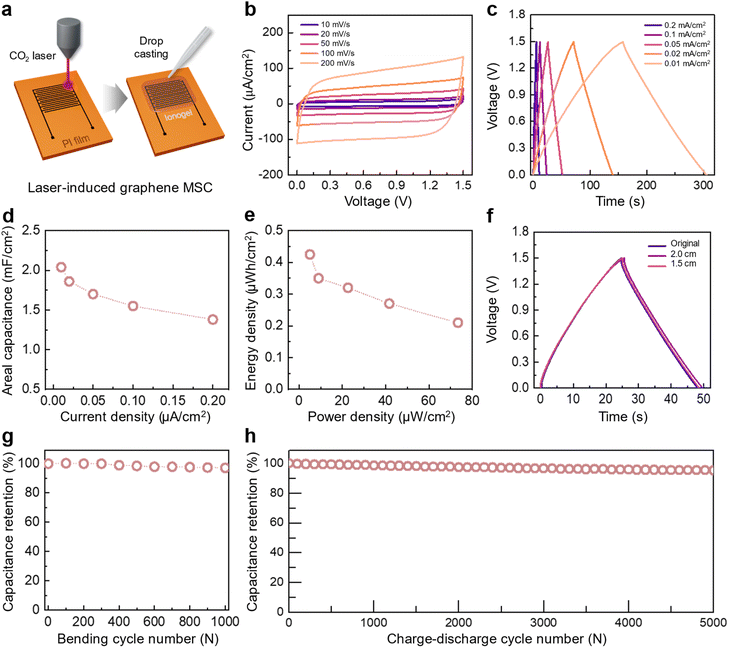 |
| | Fig. 5 Fabrication and performance of LIG-based MSCs. (a) Schematic illustration of the LIG MSCs fabrication using a CO2 laser and ionogel drop casting. (b) CV curves of the PoFPMA ionogel MSC at various scan rates at 25 °C. (c) GCD curves of the PoFPMA ionogel MSC at various current densities at 25 °C. (d) Areal capacitance of the PoFPMA ionogel MSC at various current densities. (e) Ragone plot of the PoFPMA ionogel MSC. (f) GCD curves of the PoFPMA ionogel MSC under different bending ratios. (g) Bending cycle stability of the PoFPMA ionogel MSC for 1000 cycles at a bending radius 1.5 cm. (h) Charge–discharge cycle stability of the PoFPMA ionogel MSC for 5000 cycles. | |
Low-temperature operability of the MSCs
The electrochemical responses of MSCs using PEMA, PtFEMA, and PoFPMA ionogels were compared across temperatures ranging from 20 to −30 °C. In contrast to the PoFPMA MSC, which preserved its rectangular CV curves even at −20 °C, the PEMA and PtFEMA MSCs showed nearly linear CV curves at the same temperature (Fig. S20). Temperature-dependent GCD measurements further revealed operational limits among the three MSCs. While the PEMA MSC failed to operate at 0 °C and the PtFEMA MSC failed to operate below −20 °C, the PoFPMA MSC maintained distinct charge–discharge profiles even at −30 °C (Fig. S21). The areal capacitance derived from GCD profiles, was compared across this temperature range to assess the low-temperature charge storage performance of the three MSCs (Fig. 6a). The PoFPMA MSC operated down to −30 °C, whereas the PtFEMA MSC and PEMA MSC operated only down to −10 °C and 0 °C, respectively (Fig. 6a). During the stepwise temperature decrease (15 cycles per temperature), the PoFPMA MSC maintained stable capacitance down to −20 °C, outperforming both the PEMA and PtFEMA MSCs (Fig. 6b). Upon reheating, the PoFPMA MSC exhibited the highest recovery, retaining 96% of its initial capacitance and confirming its superior thermal resilience. Under −20 °C conditions, only the PoFPMA MSC remained operational, successfully lighting an LED, whereas both the PEMA and PtFEMA MSCs failed to operate under the same conditions (Fig. 6c–e). These results demonstrate that our synergistic molecular design based ionogel can function as a reliable energy-storage device even under low-temperature conditions.
 |
| | Fig. 6 Low-temperature performance of the ionogel based MSCs (a) temperature-dependent areal capacitance of MSCs with different ionogels. (b) Cycling performance of MSCs using the PEMA, PtFEMA, and PoFPMA ionogels during temperature decrease and recovery. (c–e) Photographs of MSCs using the (c) PEMA, (d) PtFEMA, and (e) PoFPMA ionogels operating at low-temperatures. | |
Conclusions
In summary, we demonstrated that a synergistic molecular design strategy enables ionogel-based energy-storage devices to operate reliably even at low-temperatures. Accordingly, we tailored copolymer gelators that (i) incorporate fluorinated side-chains to enhance ion dissociation, thereby increasing the number of mobile ion carriers, and (ii) optimize polymer miscibility and chain length to accelerate segmental dynamics, resulting in improved ion mobility within the ionogel network. As a consequence, the PoFPMA ionogel achieved enhanced ionic conductivity and lower temperature dependence, enabling stable ion-transport even at low-temperatures. Leveraging these properties, we fabricated MSCs using the PoFPMA ionogel, which demonstrated reliable and efficient electrochemical operation at −30 °C. This molecular design approach can be broadly applied to create gel electrolytes and energy-storage devices capable of operating under extreme low-temperature conditions.
Experimental section
Materials
All reagents were obtained from commercial sources (Sigma-Aldrich) unless otherwise noted. The ionic liquid precursor, 1-butyl-3-methylimidazolium chloride ([BMI][Cl], Iolitec), and lithium bis(trifluoromethylsulfonyl)imide ([Li][TFSI], 3 M) were used to prepare [BMI][TFSI]. The synthesis was carried out through a single-step anion-exchange process, in which [BMI][Cl] was reacted with an excess amount of [Li][TFSI] in deionized water at 60 °C for 24 h. For the polymerization initiator, 2,2′-azobis(2-methylpropionitrile) (AIBN; supplied as a 12 wt% acetone solution in 100 mL), acetone was first removed using a rotary evaporator. The resulting solid was then purified by recrystallization in methanol.
Synthesis of copolymers
All copolymers were prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization, employing ethyl 2-(phenylcarbonothioylthio)-2-phenylacetate as the chain transfer agent (CTA). Prior to polymerization, the monomers were purified by removing inhibitors through a basic alumina column. For the synthesis of PS-r-PoFPMA, styrene (2.38 g, 22.85 mmol), 2,2,3,3,4,4,5,5-octafluoropentyl methacrylate (oFPMA) (38.84 g, 129.4 mmol), CTA (4.8 mg, 0.0152 mmol), and AIBN (0.5 mg, 0.0030 mmol) were combined in a two-neck flask and stirred mechanically. To maintain a consistent styrene content across all three copolymers, the feed ratio between ethyl methacrylate (EMA) and 2,2,2-trifluoroethyl methacrylate (tFEMA) was regulated based on the known reactivity ratios of styrene with methacrylate derivatives. The reaction mixture was degassed with argon at room temperature for 30 minutes and then heated in an 80 °C oil bath for 21 hours. After polymerization, the mixture was quenched by rapid cooling in liquid nitrogen, followed by precipitation into methanol. The resulting copolymers (PS-r-PEMA, PS-r-PtFEMA, PS-r-PoFPMA) were collected and dried under vacuum at 60 °C for 24 hours. For a fair comparison, all three copolymers were prepared with comparable total molecular weights and styrene molar fractions.
Preparation of ionogels
To prepare the ionogels, the 40 wt% copolymer and 60 wt% [BMI][TFSI] were dissolved in acetone and stirred at room temperature for 3 h. The resulting solution was cast onto a glass substrate under ambient conditions, and the gels formed after complete removal of the cosolvent.
Characterization of copolymer and ionogels
The mole fractions of styrene and the methacrylate derivatives in the copolymers were determined by 1H NMR spectroscopy (Bruker digital Avance Neo 400) using acetone-d6 (CD3COCD3; 99.9%) as the solvent. The number-average molecular weight (Mn) and polydispersity index (Đ) were obtained by size exclusion chromatography (SEC) (LC-20AT, Shimadzu) equipped with two PLgel 5 µm mixed-C 300 × 7.5 mm columns (Agilent Technologies) and a refractive index detector (RID-20A, Shimadzu). Calibration was performed with PS standards and tetrahydrofuran (THF, HPLC grade) as the eluent. The Tg of the copolymers was analyzed using DSC (DSC204 F1, Phoenix) over a temperature range of −120 to 100 °C, with nitrogen as the purge gas and a heating rate of 10 °C min−1. Rheological measurements of the ionogels were carried out on a rheometer (MCR 92, Anton Paar) using 8 mm parallel plates, with the frequency and strain fixed at 10 rad s−1 and 1%, respectively. For tTS analysis, frequency sweeps from 0.1 to 100 rad s−1 were performed across 0–150 °C, using a 1% strain and a reference temperature of 20 °C. FTIR (Spectrum 100, PerkinElmer) and Raman spectra (ARAMIS, Horiba Jobin Yvon, 633 nm laser) were recorded to examine the molecular interactions within the ionogels.
MSC fabrication
LIG electrodes were directly written on the 0.125 mm PI film using the computer-controlled CO2 laser cutter (xTool P2). The patterning was performed at a laser intensity of 4.95 W with a scan rate of 8.9 cm s−1, producing LIG electrode pattens with a thickness of 300 µm. The fabricated LIG electrodes were characterized using Raman spectroscopy (ARAMIS, Horiba Jobin Yvon) with a 633 nm laser and SEM (Hitachi SU8230). Next, 100 µL of the ionogel solution was drop-cast onto the LIG electrode patterns, and the cosolvent was evaporated for 3 hours. Silver paste was used to coat the ends of the electrodes for electrical contact.
Device characterization
The ionic conductivity and capacitance of the ionogels were evaluated using EIS (IM6, Zahner) over a frequency range of 10−1–106 Hz with an AC amplitude of 100 mV. Ionic conductivity was calculated from the expression σ = h/AR, where h, A, and R denote the gel thickness, electrode contact area, and bulk resistance, respectively. CV and GCD experiments were performed on the same EIS instrument to further assess the electrochemical behavior of the ionogels. The areal capacitance was determined from the discharge curves using the equation C = IΔt/SΔV, where I, Δt, S, and ΔV represent the discharging current (A), discharging time (s), area of the interdigitated electrode (cm2), and potential window (V), respectively. The energy density was calculated from the discharge profiles using the equation E = (CΔV2)/(2 × 3600), where C is the areal capacitance (F cm−2), ΔV is the potential window (V), and the factor 3600 converts the unit from J cm−2 to Wh cm−2. The power density was calculated using P = E/Δt, where E is the energy density (Wh cm−2) and Δt is the discharge time (h).
Author contributions
Daniel Sanghyun Cho: conceptualization, methodology, investigation, data curation, and writing – original draft. Ji Hye Han: conceptualization, methodology, investigation, visualization, and writing – original draft. Neul Kyum Ha: investigation, and formal analysis. Yong Min Kim: methodology, and investigation. Jin Chul Kim: project administration, funding acquisition, and writing – review & editing. Hong Chul Moon: supervision, project administration, funding acquisition, writing – original draft and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
The supporting data has been provided as part of the supplementary information (SI). Supplementary information: synthetic procedures for the copolymer gelators, 1H NMR spectra and GPC curves of PS-r-PEMA, PS-r-PtFEMA, and PS-r-PoFPMA, rheological measurements of the corresponding ionogels (storage and loss moduli, stress-relaxation behavior, and terminal relaxation times), DSC thermograms and temperature-dependent ionic conductivity analyses. Characterization of the LIG electrode includes optical image, SEM, and Raman spectroscopy. Additional analyses of the ionogel-based MSCs include extended electrochemical measurements such as CV, GCD, areal capacitance as a function of current density, Ragone plots, and low-temperature performance from −30 to 20 °C. See DOI: https://doi.org/10.1039/d5ta10237e.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2025-25441256 and RS-2025-00554260). Also, this research was supported by the Nano & Material Technology Development Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (RS-2023-00283244). This work was supported by the Korea Research Institute of Chemical Technology (KRICT) through the project ‘Next-Generation Automotive Materials and Components Development for Global Regulatory Compliance (Project No. DS2684-01)’ funded by the Ministry of Science and ICT.
References
- Y. Zhang, Y. Lu, J. Jin, M. Wu, H. Yuan, S. Zhang, K. Davey, Z. Guo and Z. Wen, Adv. Mater., 2024, 36, 2308484 CrossRef CAS PubMed.
- J. Tu, J. Pan, X. Liu, Y. Yan, Y. Shi, X. Yu and X. Jia, J. Mater. Chem. A, 2022, 10, 7036–7047 RSC.
- P. Lu, Z. Zhou, Z. Xiao, J. Lu, J. Zhang, G. Hu, W. Yan, S. Xia, S. Zhang, Z. Wang, H. Li, C. Wang, F. Wu and X. Sun, Joule, 2024, 8, 635–657 CrossRef CAS.
- Y. Chen, H. Jin, J. Zhang, Q. Wu, S. Han, A. Chen and L. Guan, J. Mater. Chem. A, 2024, 12, 18958–18967 RSC.
- J. He, L. Cao, J. Cui, G. Fu, R. Jiang, X. Xu and C. Guan, Adv. Mater., 2024, 36, 2306090 CrossRef CAS PubMed.
- N. Parvin, D. Merum, M. Kang, S. W. Joo, J. H. Jung and T. K. Mandal, J. Mater. Chem. A, 2025, 13, 24320–24386 RSC.
- Y. M. Kim, W. Y. Choi, J. H. Kwon, J. K. Lee and H. C. Moon, Chem. Mater., 2021, 33, 2683–2705 CrossRef CAS.
- H. W. Yang, D. S. Cho, J. Kang, J. H. Han, Y. M. Kim and H. C. Moon, JACS Au, 2025, 5, 4655–4668 CrossRef CAS PubMed.
- S. Y. Kim, Y. J. Jang, Y. M. Kim, J. K. Lee and H. C. Moon, Adv. Funct. Mater., 2022, 32, 2200757 CrossRef CAS.
- H. Chen, Y. Wang, D. Li, Y. Zhao, B. Wan, S. Long, Y. Huang and X. Li, ACS Appl. Polym. Mater., 2024, 6, 8449–8460 CrossRef CAS.
- X. Hou, Q. Zhang, L. Wang, G. Gao and W. Lü, ACS Appl. Mater. Interfaces, 2021, 13, 12432–12441 Search PubMed.
- H. Ma, Q. Zhao, P. Cheng, X. Geng, H. Tao, Z. Zhang, Y. Jiang, J. Ma, K. Yang, Q. Liu, H. Zhang, Z. Liang, J. Li, T. Wang, M. Xue and N. Zhu, J. Mater. Chem. A, 2024, 12, 20088–20096 RSC.
- D. Zhang, H. Chen, Y. Zhang, J. Yang, Q. Chen, J. Wu, Y. Liu, C. Zhao, Y. Tang and J. Zheng, Chem. Soc. Rev., 2025, 54, 5292–5341 RSC.
- C. Lu and X. Chen, Nano Lett., 2020, 20, 1907–1914 CrossRef CAS PubMed.
- M. Chen, W. Zhou, A. Wang, A. Huang, J. Chen, J. Xu and C.-P. Wong, J. Mater. Chem. A, 2020, 8, 6828–6841 RSC.
- Q. Rong, W. Lei, J. Huang and M. Liu, Adv. Energy Mater., 2018, 8, 1801967 Search PubMed.
- Z. Fu, D. Li, H. Liu, R. Liu, Q. Lyu, Y. Han, Y. Wang, K. Zhang, G. Chen and Y. Tian, Chem.–Eng. J., 2024, 488, 150775 CrossRef CAS.
- Y. Wang, Y. Xia, P. Xiang, Y. Dai, Y. Gao, H. Xu, J. Yu, G. Gao and K. Chen, Chem.–Eng. J., 2022, 428, 131171 CrossRef CAS.
- K. Meister, S. Strazdaite, A. L. DeVries, S. Lotze, L. L. C. Olijve, I. K. Voets and H. J. Bakker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17732 CrossRef CAS PubMed.
- Q. Wang, J. Gan, Y. Zhao, C. Yang, G. Zhu, N. Wang, C. Zhang, J. Yang and T. Zhou, EES Batteries, 2025, 1, 672–691 RSC.
- C. Yeo, S. Kang, Y.-J. Lee, S. Choo, J. Kim, S.-H. Yu, J.-W. Park, D.-J. Yoo and M. Kim, Adv. Mater., 2025, e15958 Search PubMed.
- K. Wang, M. Zhang, J. Ren, W. Wei and J. Nai, Nanoscale, 2025, 17, 11275–11292 RSC.
- M. Li, M. W. Reeder and T. Wu, Food Hydrocolloids, 2023, 145, 109127 CrossRef CAS.
- S. Zhang, K. H. Lee, J. Sun, C. D. Frisbie and T. P. Lodge, Macromolecules, 2011, 44, 8981–8989 CrossRef CAS.
- Y. M. Kim, K. S. Yu and H. C. Moon, Chem. Eng. J., 2024, 480, 147947 CrossRef CAS.
- J. H. Kwon and H. C. Moon, Adv. Funct. Mater., 2025, 35, 2503935 CrossRef CAS.
- P. O. Hama, S. B. Aziz and O. G. Abdullah, Sci. Rep., 2025, 15, 30475 CrossRef CAS PubMed.
- Q. Zhao, C. Shen, K. P. Halloran and C. M. Evans, ACS Macro Lett., 2019, 8, 658–663 CrossRef CAS PubMed.
- E. C. Fluker, S. Pathreeker and I. D. Hosein, J. Phys. Chem. C, 2023, 127, 16579–16587 CrossRef CAS PubMed.
- Y.-F. Huang, T. Gu, G. Rui, P. Shi, W. Fu, L. Chen, X. Liu, J. Zeng, B. Kang, Z. Yan, F. J. Stadler, L. Zhu, F. Kang and Y.-B. He, Energy Environ. Sci., 2021, 14, 6021–6029 RSC.
- C. Xing, M. Zhao, L. Zhao, J. You, X. Cao and Y. Li, Polym. Chem., 2013, 4, 5726–5734 RSC.
- Y. M. Kim, J. H. Kwon, S. Kim, U. H. Choi and H. C. Moon, Nat. Commun., 2022, 13, 3769 CrossRef CAS PubMed.
- J. Jaganathan and M. Sivapragasam, J. Chem. Eng., 2016, 7, 1–6 Search PubMed.
- G. Ye, X. Hong, M. He, J. Song, L. Zhu, C. Zheng, Y. Ma, Y. An, K. Shen, W. Shi, Y. Jia, M. B. Shafqat, P. Gao, D. Xia, F. Chen and Q. Pang, Adv. Mater., 2025, 37, 2417829 CrossRef CAS PubMed.
- I. Jeong, S. Kim, Y. Kim, C. Kim, J. Kang, J. H. Ha, Y. Cho, S. J. Kang, J. Ryu, J. W. Han and S. Park, Adv. Mater., 2025, 37, 2412652 CrossRef CAS PubMed.
- J. Chen, H. Zhang, M. Fang, C. Ke, S. Liu and J. Wang, ACS Energy Lett., 2023, 8, 1723–1734 CrossRef CAS.
- R. G. Ricarte and S. Shanbhag, Polym. Chem., 2024, 15, 815–846 RSC.
- I. You, D. G. Mackanic, N. Matsuhisa, J. Kang, J. Kwon, L. Beker, J. Mun, W. Suh, T. Y. Kim, J. B.-H. Tok, Z. Bao and U. Jeong, Science, 2020, 370, 961–965 CrossRef CAS PubMed.
- G. Jeanmairet, B. Rotenberg and M. Salanne, Chem. Rev., 2022, 122, 10860–10898 CrossRef CAS PubMed.
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. G. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS PubMed.
- T.-S. D. Le, H.-P. Phan, S. Kwon, S. Park, Y. Jung, J. Min, B. J. Chun, H. Yoon, S. H. Ko, S.-W. Kim and Y.-J. Kim, Adv. Funct. Mater., 2022, 32, 2205158 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *de and
Hong Chul
Moon
*de and
Hong Chul
Moon








