DOI:
10.1039/D5TA09144F
(Paper)
J. Mater. Chem. A, 2026,
14, 9645-9656
Modulating (001) plane growth in β-Ni(OH)2 precursors: a pathway to controlling lithiation kinetics and enhancing the structural integrity of LiNiO2
Received
11th November 2025
, Accepted 26th January 2026
First published on 3rd February 2026
Abstract
The rational design of β-Ni(OH)2 precursors is paramount for realizing the full potential of stoichiometric LiNiO2 (LNO) cathodes. This study demonstrates that strategically engineering the precursor's crystallographic architecture governs the synthesis kinetics, structural integrity, and ultimate electrochemical performance of LNO. By solely manipulating reactant injection intervals during co-precipitation, we modulated the preferential growth of the (001) crystal plane to create distinct β-Ni(OH)2 microstructures. The intentional restriction of (001) plane growth yielded a precursor with a high specific surface area, which significantly lowered the activation energy for lithiation. This kinetic advantage promoted a more complete and uniform phase transformation, resulting in a final LNO cathode with larger crystallites, lower lattice strain, and superior mechanical integrity. Conversely, the dense precursor derived from promoted (001) plane growth imposed a kinetic barrier, leading to incomplete reaction, residual inactive phases, and high internal strain. These profound structural differences translated directly into electrochemical performance. The LNO derived from the engineered precursor delivered higher capacity, superior rate capability, and better cycling stability, whereas its counterpart suffered from severe particle fragmentation and rapid degradation. This work establishes a direct, causal pathway from precursor-level crystallographic engineering to structurally robust, high-performance LNO cathodes.
Introduction
The development of high-nickel layered oxides (LiNixM1−xO2, where M represents other transition metals (TMs) and x ≥ 0.8) is central to advancing the energy density of next-generation lithium-ion batteries.1–3 Among these, stoichiometric LiNiO2 (LNO) stands as the parent compound, representing an ideal model system for understanding the intrinsic challenges and opportunities within this critical class of cathode materials.4–7 Achieving a well-ordered layered structure in LNO is paramount for superior electrochemical performance, yet its synthesis is notoriously sensitive to processing conditions, often resulting in undesirable cation mixing and the formation of electrochemically inactive rock-salt phases.8–12 The conventional synthesis of LNO involves the high-temperature solid-state lithiation of a Ni-based precursor, typically Ni(OH)2 prepared via co-precipitation.13,14 While extensive research has focused on tuning the bulk physical properties of these precursors, such as particle size and morphology,15–19 a fundamental understanding of how the precursor's intrinsic microstructure influences the kinetics of lithium incorporation and subsequent crystallographic evolution remains critically underdeveloped. It is hypothesized that the diffusion of lithium and the nucleation of the layered phase during calcination are not merely surface phenomena but are profoundly dictated by the precursor's crystallographic architecture, including its exposed lattice planes, surface area, and porosity. However, a systematic investigation directly linking these precursor microstructural attributes to the final structural and electrochemical properties of LNO has been conspicuously absent.
This study directly addresses this knowledge gap by demonstrating that the crystallographic evolution and ultimate performance of LNO can be strategically governed by tailoring the microstructure of the Ni(OH)2 precursor. As pristine LNO serves as the baseline for all Ni-rich compositions, our investigation intentionally excludes the effects of dopants and coatings to isolate the fundamental relationship between precursor design and cathode properties. We introduce a simple yet highly effective method to modulate the precursor's microstructure—specifically by controlling the preferential growth of the (001) crystal plane—by adjusting only the reactant injection interval during co-precipitation. This approach allows for precise control over the precursor's specific surface area and pore architecture without altering other critical synthesis parameters such as composition, pH, or temperature. By systematically correlating the engineered properties of the Ni(OH)2 precursors with the phase evolution, final crystal structure, and cell-level electrochemical performance of the resulting LNO, we provide unequivocal evidence that precursor-level microstructural engineering is a key determinant of a cathode's success. Post-cycle analyses further reinforce this link by revealing how the initial precursor design impacts long-term cycling stability and capacity retention. Our findings not only illuminate a clear pathway for optimizing LNO but also offer significant implications for the rational design of more complex, commercially relevant high-Ni cathode systems. This work underlines the critical importance of a bottom-up, precursor-centric approach to developing next-generation high-energy-density cathode materials.
Experimental
Synthesis of β-Ni(OH)2 precursors and LNO cathodes
The Ni(OH)2 precursors were synthesized via a co-precipitation method in a continuous stirred-tank reactor. The synthesis employed a 2 M NiSO4·6H2O solution (TM source), a 5 M NaOH solution (precipitant), and a 3 M NH4OH solution (chelating agent). To create precursors with distinct microstructures, two different solution injection strategies were employed while keeping the total injected volume constant over the 24 h reaction period. A low-porosity precursor, hereafter denoted as L–Ni(OH)2, was obtained via intermittent injection of the NiSO4·6H2O and NH4OH solutions at 17 s intervals. Conversely, a high-porosity precursor, H–Ni(OH)2, was synthesized using continuous injection of all solutions. Throughout the reaction, the temperature, pH, and stirring speed were rigorously maintained at 45.5 °C, 11.1, and 900 rpm, respectively, under a high-purity N2 atmosphere. The resulting precipitates were washed thoroughly with deionized water and dried in a vacuum oven at 60 °C for 12 h. To synthesize the final LNO cathodes, the dried Ni(OH)2 precursors were homogeneously mixed with LiOH·H2O at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.05. The mixture was then subjected to a two-step calcination process in a tube furnace under a high-purity O2 flow of 0.6 L min−1. The thermal profile consisted of a ramp to 500 °C at a rate of 2 °C min−1 with a 5 h dwell, followed by a second ramp to 650 °C at the same rate with a 10 h dwell. The final cathode materials derived from L–Ni(OH)2 and H–Ni(OH)2 are designated as L-LNO and H-LNO, respectively.
:1.05. The mixture was then subjected to a two-step calcination process in a tube furnace under a high-purity O2 flow of 0.6 L min−1. The thermal profile consisted of a ramp to 500 °C at a rate of 2 °C min−1 with a 5 h dwell, followed by a second ramp to 650 °C at the same rate with a 10 h dwell. The final cathode materials derived from L–Ni(OH)2 and H–Ni(OH)2 are designated as L-LNO and H-LNO, respectively.
Materials characterization
Particle morphology and cross-sections were examined using scanning electron microscopy (SEM; TESCAN, VEGA3) after preparation with a cross-polisher (JEOL, SM-09010). The internal nanostructure of the particles was investigated by transmission electron microscopy (TEM; FEI Titan, 80–300) on lamellae prepared using a focused ion beam (FIB; Hitachi, NX5000). Crystallographic analysis was performed using X-ray diffraction (XRD; Rigaku, MiniFlex600) with Cu Kα radiation, scanning over a 2θ range of 10° to 140° at 1° min−1. Rietveld refinement of the XRD patterns was performed using the TOPAS software for quantitative phase analysis. Specific surface areas were determined by the Brunauer–Emmett–Teller (BET) method from N2 adsorption/desorption isotherms measured at 77 K (BEL, Belsorp II mini). Pore size distributions were calculated using the Barrett–Joyner–Halenda (BJH) method. Thermogravimetric analysis (TGA; TA Instruments, TGA550) was employed to investigate the lithiation behavior during calcination and the thermal stability of chemically delithiated samples. The mechanical strength of the particles was measured using a micro-compression tester (Shimadzu MCT-510). Surface chemistry was characterized using X-ray photoelectron spectroscopy (XPS; VersaProbe, PHI 5000).
Chemical delithiation and residual lithium titration
Chemically delithiated samples were prepared by stirring the LNO cathode powder with NO2BF4 (oxidizing agent) in acetonitrile at a 1:2 mass ratio overnight within an argon-filled glovebox. The resulting delithiated product was thoroughly washed with acetonitrile to remove LiBF4 byproducts and subsequently dried under vacuum.20,21 The amount of residual lithium (LiOH and Li2CO3) on the cathode surface was quantified by double titration. A known mass (W) of the cathode powder was dispersed in deionized water and stirred for 30 min. The suspension was filtered, and the filtrate was titrated with a standardized 0.1 N HCl solution. Using phenolphthalein indicator, the volume of HCl required to reach the first endpoint was recorded as V1. Subsequently, methyl red indicator was added, and the solution was heated, cooled, and titrated to the second endpoint, with the consumed volume recorded as V2. The weight percentages of LiOH and Li2CO3 were calculated as follows:| |  | (1) |
| | 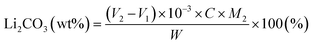 | (2) |
where C is the concentration of the HCl titrant (mol L−1), and M1 and M2 are the molecular weights of LiOH and Li2CO3 (g mol−1), respectively.22
Electrochemical measurements
Cathodes were fabricated by casting a slurry onto aluminum foil. The slurry consisted of the active material (L-LNO or H-LNO), carbon black (Super P and KS-6), and poly(vinylidene fluoride) binder in an 85:7.5:7.5 wt% ratio, dispersed in N-methyl-2-pyrrolidone. The electrodes were dried at 100 °C for 12 h, and the loading level was maintained at approximately 4.0 mg cm−2. For full-cell tests, graphite anodes were prepared from a slurry of natural graphite (POSCO Co., Ltd), carbon black (Super P and KS-6), and a styrene-butadiene rubber/sodium carboxymethyl cellulose binder (70:15:15 wt%) in deionized water. CR2032-type coin cells were assembled in an argon-filled glovebox. Half-cells comprised a cathode, a lithium metal counter electrode, and a Celgard 2400 separator. The half-cell electrolyte was 1.13 M LiPF6 in ethylene carbonate/dimethyl carbonate/diethyl carbonate (3:4:3 by vol.). Full-cells paired the LNO cathodes with the graphite anodes, with an N/P ratio of 1.1–1.2. The full-cell electrolyte was 1.2 M LiPF6 in ethylene carbonate/ethyl methyl carbonate (3:7 by vol.) with vinylene carbonate as an additive. Galvanostatic charge/discharge cycling was performed on a Wonatech WBC 3000 battery cycler at 25 °C. The voltage windows were 2.7–4.3 V (vs. Li+/Li) for half-cells and 3.0–4.2 V for full-cells. Galvanostatic intermittent titration technique (GITT) measurements were conducted at a C-rate of 0.5 C and a temperature of 25 °C. Electrochemical impedance spectroscopy (EIS) was conducted using a Wonatech ZIVE BP2 analyzer over a frequency range of 104 to 10−1 Hz. The resulting impedance spectra were fitted using an equivalent circuit model in Z-MAN software to quantify the resistive components.
Results and discussion
Synthesis of β-Ni(OH)2 precursors and LNO cathodes
In this study, we engineered β-Ni(OH)2 precursors with distinct microstructures by tailoring the coprecipitation synthesis conditions. Prior research has established that parameters such as pH can modulate the anisotropic growth of Ni-rich precursors, particularly affecting the crystallographic (001) plane.23 Enhanced growth of this plane correlates with a lower specific surface area, whereas its suppression yields a higher surface area. The (001) plane is the most thermodynamically stable facet of β-Ni(OH)2, exhibiting the lowest surface energy24 and, consequently, a slower growth rate compared to other planes.25 Leveraging these principles, we strategically designed two synthesis protocols to either promote or suppress the development of the (001) plane. For the low-porosity precursor, denoted as L–Ni(OH)2, we utilized an intermittent injection method where large volumes of TM and ammonia solutions were introduced simultaneously, followed by a 17-seconds pause. This protocol, guided by the LaMer model, generates a rapid supersaturation of monomers, leading to a brief nucleation burst followed by a prolonged crystal growth period.26–29 This extended growth phase facilitates the development of both high-energy lateral facets and the thermodynamically stable (001) plane (Fig. 1a). Conversely, the high-porosity precursor, H–Ni(OH)2, was synthesized via continuous co-injection of the reagent solutions. This approach sustains a longer nucleation phase and provides insufficient time between injections for extensive crystal growth, thereby specifically hindering the development of the slow-growing (001) plane (Fig. 1b). While SEM imaging revealed no significant difference in the secondary particle size between the two precursors (Fig. S1), their internal structures were expected to diverge.
 |
| | Fig. 1 Schematic illustrations of monomer concentration profiles during the co-precipitation of (a) L–Ni(OH)2 and (b) H–Ni(OH)2, based on the LaMer model. (c) XRD patterns of L–Ni(OH)2 and H–Ni(OH)2, with (d) a magnified view of their (001) diffraction peaks. (e) N2 adsorption/desorption isotherms and cross-sectional SEM images of (f) L–Ni(OH)2 and (g) H–Ni(OH)2. | |
To validate our synthesis strategy, we performed XRD analysis on the precursors (Fig. 1c). Both L–Ni(OH)2 and H–Ni(OH)2 were confirmed to be phase-pure β-Ni(OH)2. A magnified view of the (001) reflection revealed a distinct difference in the full width at half maximum (Fig. 1d). Using the Scherrer equation, we calculated the crystallite size along the c-axis, which corresponds to the thickness of the hexagonal plates. The vertical crystallite size was significantly larger for L–Ni(OH)2 (17.4 nm) than for H–Ni(OH)2 (11.7 nm), confirming the intended plane-selective growth. This crystallographic difference translated directly to porosity, as quantified by N2 adsorption/desorption isotherms (Fig. 1e). L–Ni(OH)2 exhibited low N2 uptake and a weak hysteresis loop, indicative of limited mesoporosity. In contrast, H–Ni(OH)2 showed substantially higher adsorption volume and a pronounced hysteresis loop, signifying a well-developed porous network. The corresponding BET specific surface area of H–Ni(OH)2 was over four times greater than that of L–Ni(OH)2 (11.33 vs. 2.47 m2 g−1). Further analysis of the pore size distribution via the BJH method (Fig. S2) showed that H–Ni(OH)2 possessed a higher total pore volume (3.083 × 10−2vs. 1.041 × 10−2 cm3 g−1) and a uniform mesoporous structure centered around 5–40 nm. Cross-sectional SEM imaging visually corroborated these findings, clearly illustrating the dense internal structure of L–Ni(OH)2versus the highly porous architecture of H–Ni(OH)2 (Fig. 1f and g). These engineered differences in porosity are hypothesized to directly influence the infiltration of molten LiOH during calcination, thereby governing the lithiation kinetics.
Impact of precursor microstructure on lithiation kinetics and phase evolution
To elucidate the influence of precursor microstructure on reaction kinetics during calcination, TGA was performed on mixtures of each precursor with a lithium source (Fig. 2a and b). This approach aligns with previous reports that employed TGA to trace lithiation pathways and phase evolution.30,31 As established by Janek and co-workers through in-depth in situ structural studies, the mass loss observed near 350 °C is not solely due to the dehydration of LiOH·H2O.32 Instead, it signifies a crucial early stage involving significant Li incorporation into the Ni sublattice to form a cubic Li1−xNixO phase (up to x = 0.3), which is essential for the subsequent formation of a uniform, near-stoichiometric LNO. Recognizing the importance of this stage, we interpret the mass loss in this temperature region as representative of the activation energy (Ea) for a series of interconnected processes, including oxygen uptake, Ni vacancy formation, and subsequent lithiation. Accordingly, the Ea for the overall reaction was determined using the Starink equation, an advanced isoconversional method.33–35 The applied Starink equation and the conversion factor (α) are defined as follows:| | 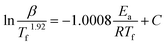 | (3) |
| | 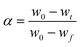 | (4) |
where β is the heating rate, Tf is the absolute final temperature, R is the gas constant, w0 is the initial sample weight, wt is the weight at a given temperature, and wf is the final weight for the region of interest. The calculated Ea values (Fig. 2c), derived from the slope of the linear fit of ln(β/Tf1.92) versus 1/Tf (Fig. S3), reveal significantly higher Ea for L–Ni(OH)2 compared to H–Ni(OH)2. This conclusion is corroborated by derivative weight-loss and differential thermal analyses in the ∼350 °C region (Fig. S4), which demonstrate that the lithiation reaction occurs at a noticeably lower temperature for H–Ni(OH)2 (341.6 °C) than for L–Ni(OH)2 (367.6 °C). Crucially, these thermal measurements were conducted at a heating rate of 2 °C min−1—identical to the rate used during cathode synthesis—ensuring that this kinetic interpretation is directly relevant to the synthesis conditions. This result supports our hypothesis that microstructural differences in the Ni(OH)2 precursors are preserved after their conversion to NiO. Specifically, the high-porosity NiO derived from H–Ni(OH)2 provides an abundance of reaction sites, facilitating Ni vacancy formation and lithiation. This hypothesis is substantiated by BET analysis, which confirms that a substantial difference in specific surface area is maintained even after conversion to NiO at 300 °C (152.28 m2 g−1 for H–Ni(OH)2-derived NiO vs. 119.86 m2 g−1 for L–Ni(OH)2-derived NiO). The corresponding N2 adsorption/desorption isotherms (Fig. S5) support this observation. These extensive internal surfaces provide more accessible sites for Li insertion into the NiO lattice, thereby reducing the Ea. The limited surface area of the L–Ni(OH)2-derived NiO consequently creates fewer reaction sites, leading to a higher Ea. This elevated energy barrier presents a significant kinetic impediment to the phase transition from NiO to the final layered LNO structure during subsequent calcination stages.
 |
| | Fig. 2 TGA curves of the (a) L–Ni(OH)2 and (b) H–Ni(OH)2 precursors mixed with a LiOH source. (c) Li insertion activation energies for L-LNO and H-LNO as a function of the conversion factor (α). (d) Evolution of crystallite sizes for L-LNO and H-LNO during the early stage of calcination. (e) Crystallite size variation of L-LNO and H-LNO with different holding times at 650 °C. (f) Grain growth rate constants for L-LNO and H-LNO, determined from a linear fit of ln(D − D0) versus lnt. | |
Given that the overall reaction rate is slower in the L–Ni(OH)2-derived material, we anticipated corresponding differences in phase evolution and grain growth. To investigate this, ex situ XRD analysis was performed at various stages of the calcination process (full patterns in Fig. S6). While no significant crystallite growth of the LNO phase was noted in the early, low-temperature stages (Fig. 2d), the analysis focused on the 500 °C step, a critical temperature that exceeds the melting point of LiOH (462 °C) and marks the onset of molten LiOH infiltration.36 Quantitative Rietveld refinement was performed on XRD data collected at 500 °C and after a 5-hours hold at this temperature to determine the relative fractions of the intermediate lithiated NiO (Li0.3Ni0.7O) and the Li-poor LNO (Li0.75Ni1.25O2) phases (Fig. S7). The refinement results indicate that H-LNO consistently contains a higher fraction of the Li-poor LNO phase than L-LNO. This disparity, already present at the 500 °C mark (51.0% vs. 44.5%), becomes more pronounced after the 5-hours hold (79.4% vs. 65.9%). This finding suggests more effective initial lithium incorporation into the H-LNO intermediate. During the subsequent hold, the higher permeability of the porous H–Ni(OH)2 precursor to molten LiOH likely enhances the solid/liquid interfacial contact area, further accelerating the phase conversion. At the higher calcination temperature of 650 °C, a distinct increase in the LNO crystallite size with holding time was observed (Fig. 2e). The grain growth rate constant (k) was calculated using the crystallite sizes (D) determined from the Scherrer equation, based on the following relationship:37–39
| | 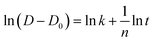 | (5) |
where
D0 is the initial crystallite size,
t is the calcination time, and
n is the grain growth exponent. The analysis reveals faster grain growth kinetics for the H–Ni(OH)
2-derived LNO (
Fig. 2f). The
k value was significantly higher for the H–Ni(OH)
2-based material (0.135 nm
2 s
−1vs. 0.097 nm
2 s
−1), confirming that its favorable precursor microstructure translates to accelerated LNO phase formation and growth.
Physicochemical and crystallographic properties of final LNO materials
The kinetic differences during synthesis manifested in distinct physicochemical properties of the final LNO powders. Cross-sectional SEM revealed that H-LNO consists of densely packed primary particles, whereas L-LNO exhibits a more loosely aggregated internal structure (Fig. 3a and b). This morphological difference conferred superior mechanical integrity to H-LNO, as confirmed by micro-compression tests (Fig. S8). The average compressive strength of H-LNO (206.48 MPa) was significantly higher than that of L-LNO (162.32 MPa), attributable to more effective stress distribution across the larger contact areas between its tightly packed primary particles. This disparity in interparticle compactness was expected to influence bulk porosity, which was subsequently investigated using N2 adsorption/desorption isotherm analysis (Fig. 3c). L-LNO exhibited a type IV isotherm with a slight H3 hysteresis loop, indicative of mesoporosity, and a higher N2 adsorption volume, corresponding to a larger specific surface area of 0.59 m2 g−1. Conversely, H-LNO displayed a type II isotherm with negligible hysteresis and a lower adsorption volume, consistent with a less porous, more compact structure, resulting in a smaller specific surface area of 0.27 m2 g−1. These results align with the SEM observations and suggest that the differences in primary particle packing and bulk porosity would influence the LNO's resistance to particle cracking and its electrolyte accessibility during electrochemical cycling. Notably, SEM analysis of the exterior of the secondary particles showed no significant difference in particle size between H-LNO and L-LNO, which is consistent with their precursor morphologies (Fig. S9).
 |
| | Fig. 3 Cross-sectional SEM images of (a) L-LNO and (b) H-LNO. (c) N2 adsorption/desorption isotherms. (d) XRD patterns and (e) corresponding W–H plots for L-LNO and H-LNO. | |
Crystallographic features were subsequently investigated using XRD, as presented in Fig. 3d. Both L-LNO and H-LNO patterns were indexed to the layered R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, with no detectable impurity phases. Building on the observation from Fig. 2e that H-LNO exhibits faster grain growth, the Williamson–Hall (W–H) equation was applied to the XRD patterns to decouple the effects of crystallite size and lattice strain.41,42 In the W–H plot (Fig. 3e), the y-intercept is inversely proportional to the crystallite size, while the slope is proportional to the inhomogeneous lattice strain. Consistent with crystallite sizes calculated via the Scherrer equation, H-LNO showed a lower y-intercept than L-LNO, confirming its larger crystallite size. Interestingly, the steeper slope observed for L-LNO indicates a higher degree of lattice strain, whereas the lower strain in H-LNO appears to facilitate more extensive crystallite growth, a finding that correlates well with its higher crystal growth rate constant. Detailed crystallographic parameters, summarized in Table 1, were extracted through Rietveld refinement of the XRD data (Fig. S10). L-LNO was found to have a larger TM slab thickness, potentially related to a decrease in Ni–O bond strength, which may deteriorate thermal stability.43 Conversely, L-LNO exhibited a narrower Li slab spacing. According to the literature, a constricted Li slab can impede Li+ diffusion pathways, thereby hindering ionic transport.40,44,45 Furthermore, the intensity ratio of the (003) to (104) reflections (I(003)/I(104)), a common indicator of cation disorder,46,47 was slightly lower for H-LNO. This suggests a marginally higher degree of Li+/Ni2+ cation mixing, which was quantified by refinement to be 2.5% for H-LNO versus 1.9% for L-LNO. This result was corroborated by XPS of the Ni 2p3/2 region (Fig. S11), which revealed a higher concentration of surface Ni2+ in H-LNO. Finally, the residual lithium content (LiOH and Li2CO3) was quantified by titration. L-LNO contained a higher amount of residual lithium (3.75%) than H-LNO (2.99%), a trend supported by O 1s XPS analysis, where the relative peak area corresponding to lithium residues was greater for L-LNO (89.7%) than for H-LNO (87.1%) (Fig. S12). This suggests that the less porous microstructure of the L–Ni(OH)2 precursor hindered complete lithiation during calcination.
m space group, with no detectable impurity phases. Building on the observation from Fig. 2e that H-LNO exhibits faster grain growth, the Williamson–Hall (W–H) equation was applied to the XRD patterns to decouple the effects of crystallite size and lattice strain.41,42 In the W–H plot (Fig. 3e), the y-intercept is inversely proportional to the crystallite size, while the slope is proportional to the inhomogeneous lattice strain. Consistent with crystallite sizes calculated via the Scherrer equation, H-LNO showed a lower y-intercept than L-LNO, confirming its larger crystallite size. Interestingly, the steeper slope observed for L-LNO indicates a higher degree of lattice strain, whereas the lower strain in H-LNO appears to facilitate more extensive crystallite growth, a finding that correlates well with its higher crystal growth rate constant. Detailed crystallographic parameters, summarized in Table 1, were extracted through Rietveld refinement of the XRD data (Fig. S10). L-LNO was found to have a larger TM slab thickness, potentially related to a decrease in Ni–O bond strength, which may deteriorate thermal stability.43 Conversely, L-LNO exhibited a narrower Li slab spacing. According to the literature, a constricted Li slab can impede Li+ diffusion pathways, thereby hindering ionic transport.40,44,45 Furthermore, the intensity ratio of the (003) to (104) reflections (I(003)/I(104)), a common indicator of cation disorder,46,47 was slightly lower for H-LNO. This suggests a marginally higher degree of Li+/Ni2+ cation mixing, which was quantified by refinement to be 2.5% for H-LNO versus 1.9% for L-LNO. This result was corroborated by XPS of the Ni 2p3/2 region (Fig. S11), which revealed a higher concentration of surface Ni2+ in H-LNO. Finally, the residual lithium content (LiOH and Li2CO3) was quantified by titration. L-LNO contained a higher amount of residual lithium (3.75%) than H-LNO (2.99%), a trend supported by O 1s XPS analysis, where the relative peak area corresponding to lithium residues was greater for L-LNO (89.7%) than for H-LNO (87.1%) (Fig. S12). This suggests that the less porous microstructure of the L–Ni(OH)2 precursor hindered complete lithiation during calcination.
Table 1 Summary of Rietveld refinement results for the L-LNO and H-LNO samplesa
| LNO |
a
hex (Å) |
c
hex (Å) |
Z
ox
|
I
(003)/I(104) |
Grain size (nm) |
Ni2+ ions in Li slab (%) |
T
TMO6 (Å) |
T
LiO6 (Å) |
The table lists the hexagonal lattice parameters (ahex and chex) and the atomic coordinate of the oxygen ions (Zox). The thicknesses of the TM slab (TTMO6) and the lithium slab (TLiO6) were calculated using the following equations:  and and 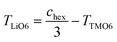 , respectively.40 , respectively.40
|
| L-LNO |
2.875 |
14.189 |
0.25158 |
1.508 |
27.8 |
1.9 |
2.319 |
2.411 |
| H-LNO |
2.876 |
14.193 |
0.25556 |
1.354 |
33.8 |
2.5 |
2.208 |
2.523 |
To probe the microstructural origins of these crystallographic differences, TEM was employed. Analysis of the particle surface regions (Fig. 4a and g) revealed distinct features. The high-angle annular dark-field (HAADF) image of L-LNO (Fig. 4b) and its corresponding line profile and fast Fourier transform (FFT) pattern (Fig. 4c) confirmed a well-ordered, conventional layered structure with a d-spacing of 2.5 Å. In stark contrast, the HAADF image of H-LNO (Fig. 4h) revealed a superlattice-like structure, indicative of Li-TM cation ordering.48 The line profile and FFT pattern (Fig. 4i) supported this observation, showing a lattice doubling characteristic of the periodic alternation of Li and TM ions.49 This phenomenon is reportedly favored when Ni3+ is reduced to Ni2+, facilitating TM migration into the Li layers.50–52 We postulate that the lower energy barrier for lithiation in H-LNO induced a localized lithium excess at the particle surface, promoting this Ni3+ reduction and subsequent superlattice formation. Finally, the bulk internal structures were examined. The high-resolution TEM (HRTEM) image of a primary particle in L-LNO (Fig. 4d) indicated the coexistence of multiple phases. At the particle surface, the FFT pattern (Fig. 4e) identified a rock-salt NiO-like phase (Fmm) that had not fully transformed into the layered structure, an observation confirmed by the inverse FFT image showing a d-spacing of 0.24 nm for the (111) plane (Fig. S13). This incomplete transformation is attributed to insufficient lithiation, likely caused by a combination of high Ea for Li incorporation into the NiO intermediate and poor penetration of molten LiOH into the low-porosity L–Ni(OH)2 precursor. In contrast, the particle interior (Fig. 4f) showed a typical layered structure. Conversely, HRTEM analysis of H-LNO particles (Fig. 4j) revealed no phase coexistence; FFT patterns from both the surface and interior (Fig. 4k and l) confirmed a well-developed and uniform layered structure throughout the particle, highlighting its more efficient and complete crystallographic transformation. Notably, the superlattice-like ordering at the H-LNO surface and the NiO-like rock-salt domains at the L-LNO surface were consistently observed across multiple particles and regions; the images presented in Fig. 4 are representative examples of these recurring structural features.
 |
| | Fig. 4 Bright-field TEM images of the surface regions of (a) L-LNO and (g) H-LNO. (b and h) HAADF-STEM images with corresponding line scan profiles from the yellow regions, and (c and i) FFT patterns from the red-dotted regions for L-LNO and H-LNO, respectively. (d and j) HRTEM images of the bulk regions of (d) L-LNO and (j) H-LNO. (e and f) and (k and l) show FFT patterns from the yellow- and blue-dotted regions for L-LNO and H-LNO, respectively. The FFT pattern in (e) is indexed to the [110] zone axis, whereas those in (f, k and l) are indexed to the [100] zone axis. | |
Electrochemical performance and degradation mechanisms
To elucidate the influence of precursor microstructure and lithiation behavior on the electrochemical performance of the resulting cathodes, a series of electrochemical analyses were conducted. The long-term cycling stability test revealed that H-LNO exhibited both a higher initial discharge capacity and superior capacity retention compared to L-LNO (Fig. 5a). The inferior initial capacity of L-LNO can be attributed to two primary factors: (i) a narrower interlayer spacing of the Li slab, which impedes Li+ diffusion,53 and (ii) the presence of an electrochemically inert NiO-like phase within the particle bulk, as confirmed by TEM analysis. Furthermore, the differential capacity (dQ/dV) plots from the initial activation cycle (Fig. S14) show that the primary oxidation peak for H-LNO occurs at a lower potential than that of L-LNO, indicating more favorable kinetics. The distinct microstructures of the two materials also profoundly impact their degradation pathways. The larger specific surface area of L-LNO, while facilitating initial electrolyte penetration,54 concurrently accelerates the propagation of microcracks during cycling. This vulnerability is likely due to the looser packing of its primary particles, which renders the structure susceptible to localized stress accumulation during volumetric changes. In contrast, the lower surface area of H-LNO inherently limits its interfacial reactivity with the electrolyte. Its more densely packed primary particles enable a more uniform distribution of internal stress during charging, thereby mitigating the formation of microcracks. In addition, the surface superlattice structure unique to H-LNO is hypothesized to act as atomic-scale pillars, reinforcing the lattice framework against collapse during deep delithiation.49,51,55,56 This inherent structural robustness accounts for the superior capacity retention of H-LNO (83.6%) over L-LNO (77.5%). When compared with previously reported LNO cathode materials (Fig. S15), L-LNO exhibits performance similar to conventional LNO, whereas H-LNO demonstrates both higher initial capacity and superior cycling stability. Rate capability tests further underscore the advantages of the H-LNO structure, which demonstrated significantly less capacity loss at higher current densities (Fig. 5b). This enhanced performance is ascribed to the wider Li slab spacing in H-LNO, which facilitates more rapid Li+ diffusion. Moreover, GITT analyses reveal that H-LNO possesses higher Li+ diffusion coefficients than L-LNO during both charging and discharging processes (Fig. S16). To validate these findings over extended operation, full-cell cycling tests were performed (Fig. 5c), confirming that H-LNO maintained higher initial capacity and better stability, consistent with the half-cell results. Analysis of the voltage profiles over 100 cycles (Fig. 5d) shows that L-LNO underwent more pronounced capacity fading (red arrow) and voltage decay (purple arrow) than H-LNO. This progressive electrochemical degradation suggests severe structural deterioration and increased charge-transfer resistance. The corresponding dQ/dV plots (Fig. 5e) reveal that the redox peaks for L-LNO gradually shift to higher potentials during charging and lower potentials during discharging, indicative of increasing polarization and hindered Li+ transport. Moreover, the overall peak intensity for L-LNO diminishes over cycling. A magnified view of the high-voltage region (Fig. S17) compellingly shows that the H2/H3 phase transition peak in L-LNO completely vanishes after 100 cycles, whereas it remains partially preserved in H-LNO. These results collectively indicate that L-LNO suffers from a more severeand irreversible structural transformation, leading to a loss of its high-voltage lithium storage capability.57–59
 |
| | Fig. 5 (a) Long-term cycling stability and (b) rate capability of L-LNO and H-LNO in half-cells. (c) Extended cycling performance of full-cells comprising L-LNO and H-LNO cathodes paired with graphite anodes over 500 cycles. (d) Voltage profiles and (e) corresponding dQ/dV plots for L-LNO and H-LNO half-cells at the 100th cycle. Nyquist plots of fresh cells and cells after 100 cycles, measured at 4.2 V vs. Li+/Li during (f) charging and (g) discharging. (h and i) RCEI values of fresh (Bef) and cycled (Aft) L-LNO and H-LNO during (h) charging and (i) discharging. (j and k) Rct values of fresh (Bef) and cycled (Aft) L-LNO and H-LNO during (j) charging and (k) discharging. | |
To further deconstruct the origins of electrochemical degradation, EIS was performed on both materials before cycling and after 100 cycles. The measurements were conducted at 4.2 V (vs. Li+/Li), a critical potential corresponding to the H2/H3 phase transition,60 to probe changes in interfacial and charge-transfer kinetics. The resulting Nyquist plots (Fig. 5f and g) were fitted using the equivalent circuit model shown in Fig. S18, which isolates the contributions of electrolyte resistance (Rs), cathode/electrolyte interphase (CEI) resistance (RCEI), and charge-transfer resistance (Rct). In the pristine state (Fig. 5h and i), L-LNO exhibited a higher RCEI value than H-LNO, a phenomenon ascribed to the formation of a thicker initial CEI layer resulting from its larger specific surface area. This disparity in RCEI persisted after 100 cycles (Fig. 5j and k). More notably, L-LNO displayed a pronounced increase in Rct, evidenced by the significantly enlarged high-to-medium frequency semicircle in its Nyquist plot. This substantial rise in Rct is a direct consequence of the aforementioned irreversible structural degradation and parasitic side reactions, confirming that the collapse of the crystalline framework in L-LNO severely impedes the electrochemical kinetics.
To validate the structural degradation inferred from the preceding electrochemical tests, we conducted post-mortem analyses on the cycled cathodes using cross-sectional SEM and XRD. Cross-sectional SEM images (Fig. 6a and b) reveal that the L-LNO cathode experienced pronounced particle fragmentation and microcracking, indicative of severe mechanical degradation. In contrast, the H-LNO cathode exhibited a remarkably intact morphology with significantly suppressed crack formation. Furthermore, XRD analysis highlighted distinct differences in the phase evolution of the two materials post-cycling. For the L-LNO cathode (Fig. 6c), the intensities of the peaks corresponding to the layered structure were substantially attenuated. The pronounced intensity loss of the (003) peak, in particular, signifies a disruption of the long-range crystallographic order,61 a direct consequence of the mechanical failure observed via SEM. Conversely, the XRD pattern of the cycled H-LNO cathode (Fig. 6d) shows well-preserved layered peaks, with the (003) peak remaining nearly intact. This result confirms the superior crystallographic and mechanical integrity of H-LNO during cycling.
 |
| | Fig. 6 Cross-sectional SEM images of (a) L-LNO and (b) H-LNO electrodes after 100 cycles. (c and d) XRD patterns of the (c) L-LNO and (d) H-LNO electrodes before and after 100 cycles. (e) TGA curves of chemically delithiated L-LNO and H-LNO. (f) Corresponding derivative weight loss curves for L-LNO and H-LNO in the temperature range of 180–250 °C. | |
To further probe the thermal and structural stability under lithium-deficient conditions, TGA was performed on chemically delithiated samples in a nitrogen atmosphere (Fig. 6e). Under this inert atmosphere, the observed weight loss is primarily attributed to lattice oxygen release driven by structural decomposition. Consistent with previous studies, the first region of major weight loss corresponds to the transition from a layered to a mixed-phase (spinel and rock-salt) structure, while the second region reflects the decomposition of the fully formed rock-salt phase.62 Notably, L-LNO exhibited a greater total weight loss than H-LNO (20.1% vs. 17.4%), indicating more severe structural collapse and a higher degree of oxygen release. Analysis of the derivative TGA curves provides more subtle details regarding their thermal behavior (Fig. 6f). The onset temperature for major decomposition was significantly lower for L-LNO (215.5 °C) than for H-LNO (229.0 °C), confirming the superior thermal stability of the latter. These findings suggest that even in a deeply delithiated state, Ni ions within the superlattice-like structure of H-LNO act as structural pillars, supporting the layered framework and mitigating its collapse. We also attribute this enhanced stability to the smaller interlayer spacing between TM slabs in H-LNO, which implies stronger TM–O bonding. This is consistent with reports showing that stronger TM–O bonds elevate the onset temperature for thermal runaway.43 Therefore, the reduced Ni–O bond length in H-LNO likely reinforces its structural integrity under thermal stress, contributing to its overall improved thermal robustness.
Conclusions
We demonstrated that the crystallographic architecture of the β-Ni(OH)2 precursor is a paramount and controllable determinant of the synthesis kinetics, final structural integrity, and ultimate electrochemical performance of LNO cathodes. By strategically manipulating only the reactant injection interval during co-precipitation, we successfully engineered two distinct precursor microstructures. Our findings reveal a clear causal relationship between this initial precursor design and the subsequent material evolution. The high porosity of H–Ni(OH)2 was shown to significantly lower the Ea for lithiation by providing extensive reaction interfaces. This kinetic advantage facilitated a more complete and uniform phase transformation into the desired layered LNO structure, resulting in a cathode characterized by larger crystallites, lower lattice strain, enhanced mechanical strength, and superior thermal stability. Crucially, this favorable kinetic pathway also induced the formation of a unique, structurally reinforcing superlattice at the particle surface. Conversely, the dense microstructure of the L–Ni(OH)2 precursor imposed a significant kinetic barrier to lithiation. This impediment led to an incomplete reaction, evidenced by the presence of an electrochemically inactive, rock-salt NiO-like phase and higher residual lithium content in the final cathode. The resulting L-LNO material suffered from higher internal strain, a constricted Li slab that hindered ionic diffusion, and poor inter-particle packing, rendering it mechanically fragile. These profound structural differences translated directly into electrochemical performance. The well-ordered H-LNO cathode delivered a higher initial capacity, superior rate capability, and outstanding cycling stability, maintaining its structural and crystallographic integrity even after extended cycling. In contrast, the L-LNO cathode exhibited rapid capacity fading and voltage decay, driven by severe particle fragmentation, microcracking, and a dramatic increase in Rct. In summary, this work provides fundamental insights into the critical role of precursor-level engineering. It establishes a clear and direct pathway from tuning precursor porosity to achieving a structurally robust and electrochemically superior high-Ni cathode. This bottom-up, precursor-centric design philosophy represents a powerful and essential strategy for the rational development of next-generation, high-energy-density lithium-ion battery materials.
Author contributions
C. H. Kim, Y. B. Choi, D. S. Kwon, and J. H. Bang conceived and designed the experiments. C. H. Kim, Y. B. Choi, D. S. Kwon, and J. Shim carried out the experiments and analyzed the results. C. H. Kim, Y. B. Choi, and J. H. Bang wrote the manuscript. All authors reviewed and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ta09144f.
Acknowledgements
This research was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (RS-2025-25428459) and by the Ministry of Education (NRF-2018R1A6A1A03024231).
Notes and references
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 Search PubMed.
- S. K. Hussain and J. H. Bang, Bull. Korean Chem. Soc., 2024, 45, 4–15 Search PubMed.
- Y. J. Kim, J. Shim, J. Kim, D. Kim and J. H. Bang, J. Energy Chem., 2026, 114, 608–617 CrossRef CAS.
- T. Ohzuku, A. Ueda and M. Nagayama, J. Electrochem. Soc., 1993, 140, 1862 CrossRef CAS.
- J. Dahn, U. von Sacken and C. Michal, Solid State Ionics, 1990, 44, 87–97 Search PubMed.
- P. Kalyani and N. Kalaiselvi, Sci. Technol. Adv. Mater., 2005, 6, 689 CrossRef CAS.
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem., Int. Ed., 2019, 58, 10434–10458 Search PubMed.
- R. A. Yuwono, F.-M. Wang, N.-L. Wu, Y.-C. Chen, H. Chen, J.-M. Chen, S.-C. Haw, J.-F. Lee, R.-K. Xie and H.-S. Sheu, Chem. Eng. J., 2023, 456, 141065 CrossRef CAS.
- T. Demuth, P. Kurzhals, S. Ahmed, F. Riewald, M. Malaki, J. Haust, A. Beyer, J. r. Janek and K. Volz, Chem. Mater., 2025, 37, 3993–4004 Search PubMed.
- S. Jeon, S. K. Hussain, J. H. Bang and J. Electrochem, Sci. Technol., 2024, 15, 161–167 CAS.
- P. Kurzhals, F. Riewald, M. Bianchini, H. Sommer, H. A. Gasteiger and J. Janek, J. Electrochem. Soc., 2021, 168, 110518 Search PubMed.
- F. Riewald, P. Kurzhals, M. Bianchini, H. Sommer, J. Janek and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 020529 CrossRef CAS.
- E. H. Lee, J. Shim and J. H. Bang, Small Methods, 2026, 10, 2500606 Search PubMed.
- U.-H. Kim, D.-W. Jun, K.-J. Park, Q. Zhang, P. Kaghazchi, D. Aurbach, D. T. Major, G. Goobes, M. Dixit and N. Leifer, Energy Environ. Sci., 2018, 11, 1271–1279 RSC.
- S. H. Lee, K. Y. Kwon, B. K. Choi and H. D. Yoo, J. Electroanal. Chem., 2022, 924, 116828 CrossRef.
- R. B. Berk, T. Beierling, L. Metzger and H. A. Gasteiger, J. Electrochem. Soc., 2023, 170, 110513 CrossRef CAS.
- K.-S. Lee, S.-T. Myung, J.-S. Moon and Y.-K. Sun, Electrochim. Acta, 2008, 53, 6033–6037 CrossRef CAS.
- Y. Koshika, H. Kaneda, S. Yoshio and Y. Furuichi, ACS Appl. Energy Mater., 2022, 5, 8169–8177 Search PubMed.
- Y. Lei, J. Ai, S. Yang, H. Jiang, C. Lai and Q. Xu, J. Alloys Compd., 2019, 797, 421–431 CrossRef CAS.
- S. Venkatraman and A. Manthiram, J. Solid State Chem., 2004, 177, 4244–4250 Search PubMed.
- C. Tian, Y. Xu, D. Nordlund, F. Lin, J. Liu, Z. Sun, Y. Liu and M. Doeff, Joule, 2018, 2, 464–477 CrossRef CAS.
- L. Li, J. Chen, H. Huang, L. Tan, L. Song, H.-H. Wu, C. Wang, Z. Zhao, H. Yi and J. Duan, ACS Appl. Mater. Interfaces, 2021, 13, 42554–42563 CrossRef CAS PubMed.
- R. B. Berk, T. Beierling, L. Metzger and H. A. Gasteiger, J. Electrochem. Soc., 2023, 170, 110530 CrossRef CAS.
- A. J. Tkalych, K. Yu and E. A. Carter, J. Phys. Chem. C, 2015, 119, 24315–24322 CrossRef CAS.
- N. Streichhan, D. Goonetilleke, H. Li, M. Soleymanibrojeni, P. W. Hoffrogge, D. Schneider, B. Nestler and W. Wenzel, Surf. Interfaces, 2024, 51, 104736 CrossRef CAS.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- V. K. L. Mer, Ind. Eng. Chem., 1952, 44, 1270–1277 CrossRef.
- N. T. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- D. T. Robb and V. Privman, Langmuir, 2008, 24, 26–35 CrossRef CAS.
- A. Tayal, P. Barai, H. Zhong, O. Kahvecioglu, X. Wang, K. Z. Pupek, L. Ma, S. N. Ehrlich, V. Srinivasan and X. Qu, Adv. Mater., 2024, 36, 2312027 CrossRef CAS.
- G. J. Lee, M. A. Abbas, M. D. Lee, J. Lee, J. Lee and J. H. Bang, Small, 2020, 16, 2002292 CrossRef CAS.
- M. Bianchini, F. Fauth, P. Hartmann, T. Brezesinski and J. Janek, J. Mater. Chem. A, 2020, 8, 1808–1820 RSC.
- M. Starink, Thermochim. Acta, 2003, 404, 163–176 CrossRef CAS.
- K. Ariyoshi, K. Yuzawa and Y. Yamada, J. Phys. Chem. C, 2020, 124, 8170–8177 Search PubMed.
- J. Shim, Y. J. Kim and J. H. Bang, Small, 2024, 20, 2400518 CrossRef CAS.
- H. Li, L. Wang, Y. Song, Z. Zhang, A. Du, Y. Tang, J. Wang and X. He, Adv. Mater., 2024, 36, 2312292 Search PubMed.
- A. Habibi, M. Jalaly, R. Rahmanifard and M. Ghorbanzadeh, New J. Chem., 2018, 42, 19026–19033 RSC.
- F. Huang, H. Zhang and J. F. Banfield, Nano Lett., 2003, 3, 373–378 Search PubMed.
- J. Shim and J. H. Bang, J. Energy Chem., 2023, 82, 56–65 CrossRef CAS.
- W. Lee, S. Muhammad, T. Kim, H. Kim, E. Lee, M. Jeong, S. Son, J. H. Ryou and W. S. Yoon, Adv. Energy Mater., 2018, 8, 1701788 CrossRef.
- A. K. Zak, W. A. Majid, M. E. Abrishami and R. Yousefi, Solid State Sci., 2011, 13, 251–256 Search PubMed.
- D. S. Kwon, E. Qamar and J. H. Bang, J. Power Sources, 2024, 620, 235267 Search PubMed.
- Z. Cui, C. Liu, F. Wang and A. Manthiram, Nat. Energy, 2025, 1–12 Search PubMed.
- S. Cui, Y. Wei, T. Liu, W. Deng, Z. Hu, Y. Su, H. Li, M. Li, H. Guo and Y. Duan, Adv. Energy Mater., 2016, 6, 1501309 Search PubMed.
- K. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS.
- W. Liu, P. Oh, X. Liu, M. J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 Search PubMed.
- H. Chen, H. Yuan, Z. Dai, S. Feng, M. Zheng, C. Zheng, J. Jin, M. Wu, X. Wu and J. Lu, Adv. Mater., 2024, 36, 2401052 CrossRef CAS.
- U.-H. Kim, J.-H. Kim, J.-Y. Hwang, H.-H. Ryu, C. S. Yoon and Y.-K. Sun, Mater. Today, 2019, 23, 26–36 Search PubMed.
- U.-H. Kim, G.-T. Park, B.-K. Son, G. W. Nam, J. Liu, L.-Y. Kuo, P. Kaghazchi, C. S. Yoon and Y.-K. Sun, Nat. Energy, 2020, 5, 860–869 CrossRef CAS.
- B. Ying, J. R. Fitzpatrick, Z. Teng, T. Chen, T. W. B. Lo, V. Siozios, C. A. Murray, H. E. Brand, S. Day and C. C. Tang, Chem. Mater., 2023, 35, 1514–1526 Search PubMed.
- H. H. Sun, U.-H. Kim, J.-H. Park, S.-W. Park, D.-H. Seo, A. Heller, C. B. Mullins, C. S. Yoon and Y.-K. Sun, Nat. Commun., 2021, 12, 6552 Search PubMed.
- U.-H. Kim, G.-T. Park, P. Conlin, N. Ashburn, K. Cho, Y.-S. Yu, D. A. Shapiro, F. Maglia, S.-J. Kim and P. Lamp, Energy Environ. Sci., 2021, 14, 1573–1583 Search PubMed.
- H. Zhou, F. Xin, B. Pei and M. S. Whittingham, ACS Energy Lett., 2019, 4, 1902–1906 CrossRef CAS.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121–130 Search PubMed.
- S. H. Ahn, D. S. Kwon, H. W. Kim and J. H. Bang, Chem. Eng. J., 2025, 168882 Search PubMed.
- G. J. Lee, M. A. Abbas and J. H. Bang, Bull. Korean Chem. Soc., 2021, 42, 934–937 Search PubMed.
- U. H. Kim, H. H. Ryu, J. H. Kim, R. Mücke, P. Kaghazchi, C. S. Yoon and Y. K. Sun, Adv. Energy Mater., 2019, 9, 1803902 Search PubMed.
- C. S. Yoon, H.-H. Ryu, G.-T. Park, J.-H. Kim, K.-H. Kim and Y.-K. Sun, J. Mater. Chem. A, 2018, 6, 4126–4132 Search PubMed.
- S. Jamil, G. Wang, L. Yang, X. Xie, S. Cao, H. Liu, B. Chang and X. Wang, J. Mater. Chem. A, 2020, 8, 21306–21316 Search PubMed.
- C. S. Yoon, D.-W. Jun, S.-T. Myung and Y.-K. Sun, ACS Energy Lett., 2017, 2, 1150–1155 Search PubMed.
- Z. Cui, Q. Xie and A. Manthiram, ACS Appl. Mater. Interfaces, 2021, 13, 15324–15332 Search PubMed.
- H. J. Noh, S. T. Myung, H. G. Jung, H. Yashiro, K. Amine and Y. K. Sun, Adv. Funct. Mater., 2013, 23, 1028–1036 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab








 and
and  , respectively.40
, respectively.40


